Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Hematology
Inherited thrombocytopenias (IT) are a group of hereditary disorders characterized by a reduced platelet count sometimes associated with abnormal platelet function, which can lead to bleeding but also to syndromic manifestations and predispositions to other disorders.
- inherited thrombocytopenias
- platelets
- bleeding
1. Introduction
Platelets, or thrombocytes, are small and anuclear blood cells with discoid shape and a size of 1.5–3.0 µm which play a crucial function in primary hemostasis. Their normal life span is 9–10 days and total circulating mass 1012, thus about 1011 platelets are released each day from their bone marrow precursors, megakaryocytes, to maintain a normal circulating platelet count of 1.5 to 4 × 109/L.
Inherited thrombocytopenias (ITs) are a heterogeneous group of congenital disorders characterized by a reduction of platelet number, a widely-variable bleeding diathesis, sometimes aggravated by associated impairment of platelet function, and frequently associated with additional defects, which may heavily impact patient lives.
ITs are rare diseases, with an estimated prevalence of 2.7 in 100,000 [1] although this figure is probably underestimated because they are often misdiagnosed as immune thrombocytopenia (ITP). A recent study on the assessment of the frequency of naturally occurring loss-of-function variants in genes associated with platelet disorders (52% of which were associated with ITs) from a large genome aggregation database showed that 0.329% of subjects in the general population have a clinically meaningful loss-of-function variant in a platelet-associated gene [2].
The first IT, Bernard Soulier syndrome, was described in 1948 and subsequently only few additional forms were reported until Sanger sequencing first, and next generation sequencing later became widely applied rapidly bringing the known ITs from less than a dozen to currently at least 41 disorders caused by mutations in 42 different genes [3,4].
2. Hereditary Disorders of Platelet Number
Given the wide heterogeneity of IT, there is no consensus on their classification, and several criteria have been proposed, such as on clinical features (e.g., age at presentation, severity, associated developmental abnormalities), platelet size or inheritance pattern (e.g., autosomal dominant, autosomal recessive and X-linked) [4,10,11].
Here we have grouped them according to the pathogenic mechanisms of thrombocytopenia.
ITs are primarily caused by mutations in genes involved in megakaryocyte differentiation, maturation and platelet production [12] (Table 1, Figure 1).
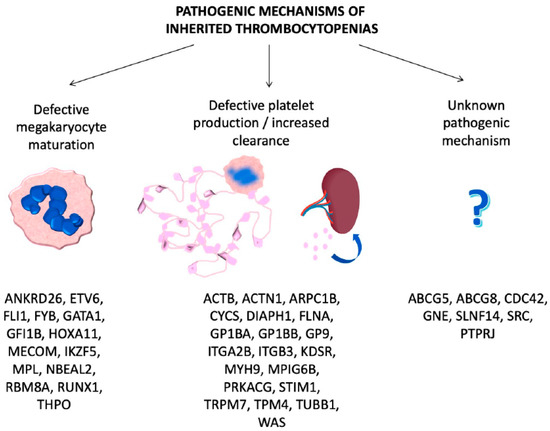
Figure 1. Genes involved in inherited thrombocytopenias classified according to the pathogenic mechanisms.
Table 1. IT classified based on the defective step of platelet count regulation involved.
| Defective Step of Thrombopoiesis | Affected Gene | Disorder | Pathogenic Mechanism (Reference) | Additional Features (e.g., Syndromic Manifestations, Predisposition) |
|---|---|---|---|---|
| Defective megakaryocyte maturation | ANKRD26 | ANKRD26-related thrombocytopenia | Loss of ANKRD26 silencing during the last phases of megakaryocytopoiesis causes ERK1/2 phosphorylation that interferes with megakaryocyte maturation [13] | Predisposition to hematological malignancies |
| ETV6 | ETV6-related thrombocytopenia | ETV6 is a transcriptional repressor that promotes the late phases of megakaryopoiesis. Mutations in ETV6 cause defective megakaryocyte maturation and impaired proplatelet formation [14] | Predisposition to hematological malignancies | |
| FLI1 | FLI1-related thrombocytopenia | FLI1 is a transcription factor regulating many genes associated with megakaryocyte development. Therefore, FLI1 mutations promote defective megakaryocyte maturation [15] | Not reported | |
| FLI1 deletion | Paris-Trousseau syndrome/Jacobsen syndrome | Abnormalities of heart and face, intellectual disabilities | ||
| FYB | FYB-related thrombocytopenia | ADAP is a protein involved in the remodeling of cytoskeleton. Mutations in ADAP cause defective maturation of megakaryocytes and clearance of platelets [16] | Mild iron deficiency anemia | |
| GATA1 | GATA1-relate disease | GATA1 is a transcription factor regulating many genes associated with megakaryocyte development therefore GATA1 defects cause alterations of megakaryocyte maturation [17] | Dyserythropoietic anemia, beta-thalassemia, congenital erythropoietic porphyria, splenomegaly | |
| GFI1B | GFI1B-related thrombocytopenia | GFI1B is a transcription factor involved in homeostasis of hematopoietic stem cells and development of megakaryocytes therefore GFI1B defects cause alterations of megakaryocyte maturation [18] | Mild myelofibrosis | |
| HOXA11 | Amegakaryocytic thrombocytopenia with radio-ulnar synostosis | HOXA11 is a transcription factor involved in the regulation of early hematopoiesis, its defect causes reduced number of megakaryocytes [19] | Bilateral radioulnar synostosis, severe bone marrow failure culminating in aplastic anemia in majority of cases, cardiac and renal malformations, hearing loss, clinodactyly, skeletal abnormalities, pancytopenia |
|
| MECOM | MECOM is a transcription factor involved in the regulation of early hematopoiesis, its defect causes reduced number of megakaryocytes [20] | |||
| IKZF5 | IKZF5-related thrombocytopenia | IKZF5 is a previously unknown transcriptional regulator of megakaryopoiesis [21] |
Not reported | |
| MPL | Congenital amegakaryocytic thrombocytopenia | MPL is the receptor for thrombopoietin. MPL defects cause impaired thrombopoietin binding and thus impaired megakaryocyte maturation [22] | Acquired bone marrow aplasia | |
| NBEAL2 | Gray platelet syndrome | Mutations in NBEAL2 cause impaired megakaryocyte maturation however its role in megakaryocytopoiesis is not clear [23] | Myelofibrosis, immune dysregulation (autoimmune diseases, positive autoantibodies, reduced leukocyte counts), proinflammatory profile | |
| RBM8A | Thrombocytopenia-absent radius | RBM8A is a protein of the exon-junction complex involved in RNA processing. It has been hypothesized that RBM8A defects cause wrong mRNA processing of unknown components of the TPO-MPL pathway impairing megakaryocyte maturation [24] | Bilateral radial aplasia, anemia, skeletal, urogenital, kidney, heart defects | |
| RUNX1 | Familial platelet disorder with predisposition to hematological malignancies | RUNX1 is a transcription factor regulating many genes associated with megakaryocyte development therefore RUNX1 mutations promote defective megakaryocyte maturation [25] | Predisposition to hematological malignancies | |
| THPO | THPO-related disease | THPO is the gene for thrombopoietin, essential for hematopoietic stem cell survival and megakaryocyte maturation [26] | Bone marrow aplasia | |
| Defective platelet production/increased clearance | ACTB | Baraitser–Winter syndrome 1 with macrothrombocytopenia | Mutations in β-cytoplasmic actin inhibit the final stages of platelet maturation by compromising microtubule organization [27] | Microcephaly, facial anomalies, mild intellectual disability, developmental delay |
| ACTN1 | ACTN1-related thrombocytopenia | ACTN-1 is involved in cytoskeletal remodeling, defects in ACTN-1 cause defective proplatelet formation [28] | Not reported | |
| ARPC1B | Platelet abnormalities with eosinophilia and immune-mediated inflammatory disease | The actin-related protein 2/3 complex (Arp2/3) is a regulator of the actin cytoskeleton and its mutation causes impaired proplatelet formation [29] | Immunodeficiency, systemic inflammation, vasculitis, inflammatory colitis, eosinophilia, eczema, lymphadenomegaly, hepato-splenomegaly, growth failure | |
| CYCS | CYCS-related thrombocytopenia | CYCS is a mitochondrial protein with a role in respiration and apoptosis. Mutations in CYCS cause ectopic premature proplatelet formation with an unknown mechanism [30] | Not reported | |
| DIAPH1 | DIAPH1-related thrombocytopenia | DIAPH1 is involved in cytoskeletal remodeling, defects in DIAPH1 cause defective proplatelet formation [31] | Hearing loss | |
| FLNA | FLNA-related thrombocytopenia | Filamin A is involved in cytoskeletal remodeling, defects in FLNA cause defective proplatelet formation [32] | Periventricular nodular heterotopia and otopalatodigital syndrome spectrum of disorders | |
| GP1BA, GP1BB, GP9 (loss of function) |
Bernard–Soulier syndrome monoallelic | The intracellular portion of the GPIb/IX/V complex links the receptor to the cytoskeleton. Disruption of this link causes impaired proplatelet formation [33] | Not reported | |
| Bernard–Soulier syndrome biallelic | ||||
| GP1BA (gain of function) |
Platelet-type von Willebrand disease | The extracellular portion of the GPIb/IX/V complex binds VWF. Constitutive binding of VWF to its receptor triggers the Src kinases pathway causing impaired proplatelet formation, ectopic platelet production and increased platelet clearance [34] | Not reported | |
| ITGA2B, ITGB3 | ITGA2B/ITGB3-related thrombocytopenia | Constitutive activation of αIIbβ3 causes cytoskeletal perturbation leading to impaired proplatelet formation [35,36] | Not reported | |
| KDSR | Thrombocytopenia and erythrokeraderma | KDSR is an essential enzyme for de novo sphingolipid synthesis, this suggests an important role for sphingolipids as regulators of cytoskeletal organization during megakaryopoiesis and proplatelet formation [37] | Dermatologic involvement ranging from hyperkeratosis/ erythema to ichthyosis. One family with no or very mild skin lesions but associated anemia has been reported |
|
| MYH9 | MYH9-related disorder | MYH9 regulates cytoskeleton remodeling and mediates signal transduction pathways involved in proplatelet formation. Abnormalities of MYH9 cause hyperactivation of the Rho/ROCK pathway causing ectopic platelet formation [38] | Kidney disease, cataract, deafness, elevated liver enzymes | |
| MPIG6B | Thrombocytopenia, anemia and myelofibrosis | G6b-B is a transmembrane receptor with an ITIM motif with a not well defined role in proplatelet formation [39] | Microcitic anemia, myelofibrosis, leukocytosis may be present |
|
| PRKACG | PRKACG-related thrombocytopenia | PKA activates many proteins involved in megakaryocyte and platelet function, among them FLNa and GPIbβ therefore its dysfunction causes impaired proplatelet formation [40] | Not reported | |
| STIM1 | Stormorken syndrome | STIM1 mutations cause a constitutively active store operated Ca2+ release-activated Ca2+ (CRAC) channel which triggers Ca2+ entry with consequent increased clearance of activated platelets [41] | Tubular myopathy and congenital myosis. Severe immune dysfunction | |
| TRPM7 | TRPM7-related thrombocytopenia | Defects of the Mg2+ channel TRPM7, a regulator of embryonic development and cell survival, cause cytoskeletal alterations resulting in impaired proplatelet formation [42] | Atrial fibrillation | |
| TPM4 | TPM4-related thrombocytopenia | Tropomyosin 4 is an actin cytoskeletal regulator. Insufficient TPM4 expression in human and mouse megakaryocytes resulted in a defect in the terminal stages of platelet production [43] |
Not reported | |
| TUBB1 | TUBB1-related thrombocytopenia | Tubulin beta1 is a major component of microtubules therefore defects in TUBB1 cause impaired proplatelet formation [44] | Not reported | |
| WAS | Wiskott–Aldrich syndrome | The WASP protein is a regulator of the actin cytoskeleton and its defect causes ectopic platelet formation and increased platelet clearance [45] | Immunodeficiency, hematopoietic malignancies, eczema, autoimmune hemolytic anemia. |
|
| X-linked thrombocytopenia | Not reported | |||
| Other/unknown pathogenic mechanism | ABCG5, ABCG8 | Thrombocytopenia associated with sitosterolemia | ABCG5 and ABCG8 regulate plant sterol and cholesterol absorption. It is supposed that sterol-enriched platelets are more rapidly cleared [46] | Xanthomas and pre-mature coronary atherosclerosis due to hypercholesterolemia |
| CDC42 | Takenouchi-Kosaki syndrome with macrothrombocytopenia |
CDC42 is a critical molecule in various biological processes including the cell cycle, cell division, and the formation of the actin cytoskeleton [47] | Defective growth and psychomotor development, intellectual disability, facial abnormalities, brain malformation, muscle tone abnormalities, immunodeficiency, eczema, hearing/visual disability, lymphedema, cardiac, genitourinary, and/or skeletal malformations |
|
| GNE | GNE-related thrombocytopenia | GNE encodes an enzyme involved in the sialic acid biosynthesis pathway and it is known that thrombocytopenia is associated with increased platelet desialylation [48] | Some patients presented myopathy with rimmed vacuoles with onset in early adulthood |
|
| SLNF14 | SLNF14-related thrombocytopenia | SLNF14 is an endoribonuclease and its role in the generation of thrombocytopenia is unknown [49] | Not reported | |
| SRC | SRC-related thrombocytopenia | Src-family kinase regulates multiple signaling pathways, its role in the generation of thrombocytopenia is unknown [50] | Myelofibrosis, bone pathologies, bone marrow dysplasia, splenomegaly, congenital facial dysmorphism |
|
| PTPRJ | PTPRJ-related thrombocytopenia | PTPRJ is a protein tyrosine phosphatase expressed abundantly in platelets and megakaryocytes, its role in the generation of thrombocytopenia is unknown [51] | None |
2.1. ITs Caused by Defective Megakaryocyte Maturation and Differentiation
ITs due to defective differentiation of hematopoietic stem cells (HSCs) into MKs are characterized by the absence or severe reduction in the number of bone marrow MKs.
ITs caused by altered MK maturation are characterized by a normal or increased number of bone marrow MKs which however are immature, dysmorphic and dysfunctional and include at least 14 different forms. Eight of these are caused by mutations of transcription factors with a key role in megakaryopoiesis, i.e., RUNX1, FLI1, GATA1, GFI1b, ETV6, HOXA11, MECOM, IKZF5. These transcription factors regulate, as activator or repressor, the expression of numerous genes, therefore these disorders are characterized by the concurrent alterations of multiple steps in MK and platelet development. For instance, RUNX1 transactivates transcription factors involved in MK maturation, proteins of the MK cytoskeleton (MYH9, MYL9, MYH10) or implicated in α and dense granule development (RAB1B, PLDN, NFE2) and members of the MK/platelet signaling pathways (ANKRD26, MPL, PRKCQ, ALOX12, PCTP) [17]. FLI1 activates the transcription of several genes associated with the production of mature MKs, including MPL, ITGA2B, GP9, GPIBA and PF4 [52]. Thrombocytopenia of TCPT/JBS, caused by deletions of the long arm of chromosome 11q, is due to reduced expression of FLI1 which is included in the deleted region.
Disorders caused by GATA1 and GFI1B mutations are associated with erythrocyte abnormalities showing the essential role of these transcription factors in controlling also red cell production. Moreover, the predisposition to haematological neoplasms of patients with RUNX1 and ETV6 variants highlights how these pathogenic variants also disrupt the homeostasis of myeloid and multipotent progenitors, respectively. Amegakaryocytic thrombocytopenia with radio-ulnar synostosis (ATRUS), a rare IT which often evolves in trilinear bone marrow failure, is due to variants in HOXA11 and MECOM, members of a family of genes encoding for DNA-binding proteins involved in the regulation of early hematopoiesis [53]. IKZF5 is a transcription factor with a non-clear role in hematopoiesis and is involved in IKZF5-RT [21].
Variants in THPO, the gene coding for thrombopoietin, a growth factor essential for hematopoietic stem cell survival and megakaryocyte maturation, and in MPL, coding for the thrombopoietin receptor, cause THPO-related thrombocytopenia (THPO-RT) and congenital amegakaryocytic thrombocytopenia (CAMT), respectively. FYB-RT is caused by variants in the FYB gene, coding for a cytoskeletal protein [16], and thrombocytopenia-absent radius is caused by variants in RBM8A, a protein of the exon-junction complex [24]. Finally, ANKRD26 and NBEAL2, proteins with an unknown role, are involved in ANKRD26-RT [13] and Gray platelet syndrome (GPS) [23], respectively.
2.2. ITs Caused by Defective Platelet Production/Clearance
ITs derived from defects of the generation of proplatelets from mature MKs and/or of the conversion of proplatelets to platelets in the bloodstream are characterized by normal MK differentiation and maturation but by ectopic release of platelets in the bone marrow and/or increased clearance of platelets from the circulation. Most of these forms are associated with enlarged platelets and derive from mutations in genes encoding for components of the acto-myosin or microtubular cytoskeletal system, such as MYH9, ACTN1, FLNA, TPM4, TRPM7 or TUBB1, or from mutations of genes for the major membrane glycoprotein (GP) complexes GPIb/IX/V and GPIIb/IIIa that indirectly affect cytoskeletal structure or reorganization, i.e., like biallelic and monoallelic Bernard Soulier syndrome (BSS) and ITGA2B/ITGB3-RT. In the latter case macrothrombocytopenia results from the disruption of the interactions of integrins with the actomyosin cytoskeleton which is essential for preserving MK cytoskeletal structure and organization. For instance, ITGA2B/ITGB3-RT is due to gain-of-function variants resulting in the constitutive, inappropriate activation of GPIIb/IIIa triggering outside-in signaling with consequent altered remodeling of the actin cytoskeleton [35,54,55]. Another example is platelet type VWD, or pseudo von Willebrand, due to gain-of-function mutations that increase the affinity of GPIbα for VWF with the consequent triggering of the Src kinases pathway downstream of activated GPIbα [34].
Wiskott–Aldrich syndrome (WAS) is a syndromic IT and X-linked thrombocytopenia (XLT) is a milder variant with only isolated thrombocytopenia which derive from mutations in the WAS gene leading to defective expression or activity of its product WASp. WASp is expressed exclusively in hematopoietic cells and has a key role in actin polymerization and cytoskeleton rearrangement. Studies in mice have shown ineffective platelet production with ectopic proplatelet formation (PPF) within the bone marrow and impaired SDF1-driven MK migration to the vascular niche [56]. The observation that splenectomy enhances the platelet count in WAS and XLT patients however, suggests that increased platelet clearance is also an important mechanism of thrombocytopenia in these disorders.
An additional group of IT belonging to those caused by impaired platelet production is due to variants in genes not directly involved in proplatelet formation, such as CYCS-RT, caused by dysfunction of a mitochondrial protein that causes thrombocytopenia by enhancing an apoptotic pathway [30], or PRKACG-RT, leading to dysfunction of PKA, which activates many proteins involved in megakaryocyte and platelet function such as FLNa and GPIbβ [40]. The Stormorken syndrome is due to gain of function mutations of STIM1 [57]. In these patients platelets circulate in an activated state due to a constitutively active store operated Ca2+ release-activated Ca2+ (CRAC) channel which triggers Ca2+ entry with consequent increased clearance of activated platelets by the spleen which causes a reduction in the number of circulating platelets [58].
Ectopic proplatelet formation in bone marrow is a peculiar mechanism causing thrombocytopenia in FYB, GP1BA (gain-of-function variants) and MYH9.
2.3. ITs Caused by Unknown Pathogenic Mechanisms
One last group of ITs is caused by variants in genes not known to be involved in megakaryocyte maturation or platelet production, and that cause thrombocytopenia by still unknown mechanisms.
An interesting IT is thrombocytopenia associated with sitosterolemia, a rare autosomal recessive disorder caused by mutations in two adjacent ATP-binding cassette transport genes (ABCG5 and ABCG8) encoding proteins (sterolins-1 and -2) that pump sterols out of cells [59]. Among the manifestations of this complex disorder due to the accumulation of sterols in plasma and cell membranes are haematological abnormalities, including thrombocytopenia, provoked by the increased stiffness of sterol-enriched membranes with possible enhanced susceptibility to lysis and rupture [60].
Another recently discovered gene causing IT is SLFN14, an endoribonuclease degrading mRNA [49,61,62]. Alongside reduced platelet number, these patients show increased platelet clearance and platelet dysfunction. However, the mechanism through which mutations in SLFN14 induce enhanced platelet turnover and abnormal platelet function is unknown. Similarly, the pathogenic mechanisms of one of the most recently reported causative genes of IT, GNE, are unknown. Mutations of GNE, the gene encoding Glucosamine (UDP-NAcetyl)-2-Epimerase/N-Acetylmannosamine kinase, cause sialuria and hereditary inclusion body myopathy [63] but are also associated with severe thrombocytopenia characterized by shortened platelet lifespan, but the exact mechanisms have not been clarified [48].
3. Diagnostic Approach
3.1. Introduction
Patients referred for investigation of bleeding symptoms should undergo preliminary laboratory investigations including full blood count, prothrombin time, activated partial thromboplastin time and von Willebrand factor (VWF) screening tests (VWF antigen, ristocetin cofactor activity and factor VIII coagulant activity). If from full blood count thrombocytopenia is identified, a diagnostic work-up for IT should be pursued. If these are normal the presence of an inherited platelet function disorder (IPFD) should be explored. IPFD are listed under Table 2.
Table 2. Inherited platelet function disorders: disorders in which platelet dysfunction is the dominant phenotypic feature independent of platelet count.
| Disease | Inheritance | Gene | Bleeding Diathesis |
|---|---|---|---|
| Arthrogryposis, renal dysfunction and cholestasis | AR | VPS33B VIPAS39 |
Severe |
| CalDAG-GEFI related platelet disorder | AR | RASGRP2 | Moderate-severe |
| Cediak-Higashi Syndrome | AR | CHS1 | Moderate-severe |
| Combined alpha-delta granule deficiency | AR/AD | Unknown | Mild-moderate |
| COX-1 deficiency | AR/AD | PTGSA | Moderate-severe |
| Delta granule deficiency | AR/AD | Unknown | Mild-moderate |
| Glanzmann thrombasthenia | AR | ITGA2B, ITGB3 | Moderate-severe |
| Glycoprotein IV (GPIV) deficiency | AR | GP4 | Mild |
| Glycoprotein VI (GPVI) deficiency | AR | GP6 | Mild |
| Gs platelet defect | AD (if paternally inherited) | GNAS | Mild |
| Hermansky–Pudlak syndrome | AR | HPS1, ADTB3A, HPS3, HPS4, HPS5, HPS6, DTNBP1, BLOC1S3, AP3D1, BLOC1S6 | Moderate-severe |
| Leukocyte adhesion deficiency, type III | AR | FERMT3 | Moderate-severe |
| P2Y12 deficiency | AR | P2RY12 | Moderate-severe |
| Phospholipase A2 (cPLA2) deficiency | not determined | PLA2G4A | Moderate-severe |
| PKCδ deficiency | AR | PRKCD | Absent |
| Primary secretion defect | AR/AD | Unknown | Mild-moderate |
| Quebec platelet disorder | AD | PLAU | Moderate-severe |
| Scott syndrome | AR | TMEM16F | Mild-moderate |
| Thromboxane A2 receptor defect | AD | TBXA2R | Mild |
| Tx synthase deficiency | AD/AR | TBXAS1 | Moderate |
The diagnostic approach to ITs can be divided into two steps. The first is the recognition of the hereditary nature of thrombocytopenia, the second is the diagnosis of a specific disorder. In fact, ITs are often confused with acquired thrombocytopenias, leading many patients to receive futile and often dangerous treatments. Careful medical history and accurate evaluation of some simple laboratory parameters help to avoid misdiagnosis [64,65]. A diagnostic algorithm for inherited thrombocytopenias was proposed several years ago and it is still valid to orient towards specific disorders [66,67]. History and clinical examination are crucial for patients with syndromic forms, whereas cell counting and the examination of peripheral blood films may guide diagnosis in non-syndromic forms [68]. However, in most cases genetic studies are required to confirm the diagnostic suspicion [3,69]. Here we propose a diagnostic flow chart for diagnosis of IT.
3.2. Clinical Examination
The first step for IT diagnosis is a careful clinical evaluation of the proband, including the personal and family bleeding history. Treatment with drugs (continuous or intermittent), recent infection, previously diagnosed haematologic disease, nonhaematologic diseases known to decrease platelet counts (e.g., eclampsia, sepsis, DIC, anaphylactic shock, hypothermia, massive transfusions), recent live virus vaccination, poor nutritional status, pregnancy, recent organ transplantation from a donor sensitized to platelet alloantigens and recent transfusion of a platelet-containing product in an allosensitized recipient should be excluded. Thrombocytopenia and/or bleeding history in other family members support the hypothesis of an IT, however a negative family history does not exclude it because some forms are recessive or derive from de novo mutations.
The most severe ITs, such as congenital amegakaryocytic thrombocytopenia or biallelic BSS, are typically identified early in infancy because of bleeding diathesis, while for several ITs spontaneous bleeding is absent or very mild explaining why they are often recognized in adult life.
Besides hemorrhagic manifestations, physical examination should also explore other organs/systems abnormalities for syndromic ITs.
In most syndromic forms the associated manifestations are present since the first months of life, such as in CAMT, Jacobsen and Wiskott–Aldrich syndrome and thrombocytopenia with absent radii, while in others they may become apparent later in life, such as renal failure in MYH9-RD, and in the latter case their genetic origin may be missed.
3.3. Laboratory Tests
At the first identification of thrombocytopenia, “pseudothrombocytopenia”, a relatively common artifactual phenomenon caused by platelet clumping in the test tube due to the presence of EDTA (ethylenediaminetetraacetic acid) as anticoagulant accounting for 0.07% to 0.27% of all cases of isolated thrombocytopenia, should be excluded [70].
Evaluation of peripheral blood smears can guide the diagnostic workup because 29 of the 41 forms that have been identified so far display morphological abnormalities of platelets, granulocytes, and/or erythrocytes [68].
When platelet size is reduced, X-linked thrombocytopenia (XLT), WAS and ITs associated with variants in FYB and PTPRJ should be considered [71]. When platelet size is enhanced MYH9-RD, BSS, GPS, thrombocytopenia linked to DIAPH1, FLNA, GATA-1, GNE, TUBB-1, GFI1b, PRKACG, SLF14, TRPM7, TPM4 and ACTN1, Paris-Trousseau thrombocytopenia, PT-VWD, ITGA2B/ITGB3-RT or thrombocytopenia associated with sitosterolemia should be considered. Among these, giant platelets characterize MYH9-RD, bBSS and TUBB1-RT. ITs associated with a normal platelet size instead are ATRUS, SRC-RT, TAR, thrombocytopenia and erythrokeraderma, CYCS-RT, FLI1-RT, IKZF5-RT, THPO-RT, ANKRD26-RT, CAMT, ETV6-RT and FPD/AML.
Abnormality of platelet granules may be observed in some ITs, with reduced or absent granules with enlarged platelets in GPS and GFI1b-RT and with reduced granules with normal-sized platelets in ANKRD26-RT [13,23,72].
Immunofluorescence performed on blood smears has recently been proposed as a method to identify defective membrane protein expression, disturbed distribution of cytoskeletal proteins, and reduction of α or delta granules, however this method requires interlaboratory validation [68].
Classic tests of platelet function, such as aggregometry (light transmission or impedance aggregometry), flow cytometry, secretion assays, electron microscopy and western blotting, may help for some ITs as subsequent steps in the diagnostic algorithm (Table 3) [18,73,74,75].
Table 3. Main features of inherited thrombocytopenias.
| Form | Disease | Inheritance | Degree of Thrombocytopenia | Key Laboratory Features | References |
|---|---|---|---|---|---|
| Syndromic | Amegakaryocytic thrombocytopenia with radio-ulnar synostosis (ATRUS) | AD | severe | Normal platelet size and morphology | [19,20] |
| Baraitser–Winter syndrome 1 with macrothrombocytopenia | AD | absent | Macrothrombocytopenia; leukocytosis with eosinophilia, leukopenia | [27] | |
| FLNA-related thrombocytopenia | XL | moderate | Macrothrombocytopenia; impaired platelet aggregation GPVI-triggered; heterogeneous α-granules, occasionally giant; abnormal distribution of FLNa | [32] | |
| GATA-1-related disease | XL | severe | Macrothrombocytopenia; reduced platelet aggregation by collagen and ristocetin; reduced α-granule content and release | [17] | |
| GNE-related thrombocytopenia | AR | from mild to severe | Macrothrombocytopenia | [48] | |
| Gray platelet syndrome | AR | moderate/severe | Macrothrombocytopenia; grey or pale platelets; dyserytropoiesis; absence of α-granules; defective TRAP-induced platelet aggregation | [23] | |
| Paris-Trousseau thrombocytopenia, Jacobsen syndrome | AD | severe | Macrothrombocytopenia; defective platelet aggregation by thrombin; giant α-granules | [15] | |
| Platelet abnormalities with eosinophilia and immune-mediated inflammatory disease | AR | moderate | Small platelets; eosinophilia; reduced platelet spreading; decreased platelet dense granules | [29] | |
| PTPRJ-related thrombocytopenia | AR | moderate/severe | Microthrombocytopenia; impaired activation by the GPVI-specific agonist convulxin and the thrombin receptor-activating peptide but normal response to ADP | [51] | |
| SRC-related thrombocytopenia | AD | moderate/severe | Platelets deficient in granules and rich in vacuoles | [50] | |
| Stormorken syndrome | AD | moderate/severe | Howell-Jolly bodies in red blood cells; enhanced annexin V binding, defective GPIIb/IIIa activation (PAC-1) | [41] | |
| Takenouchi-Kosaki syndrome with macrothrombocytopenia |
AD | absent | Macrothrombocytopenia, abnormal platelet spreading and filopodia formation | [47] | |
| Thrombocytopenia-absent radius syndrome (TAR) | AR | severe | Normal platelet size and morphology, thrombocytopenia | [24] | |
| Thrombocytopenia and erythrokeraderma | AR | moderate | Thrombocytopenia and presence of 3-keto-dihydrosphingosine in plasma | [37] | |
| Thrombocytopenia, anemia and myelofibrosis | AR | mild/moderate | Macrothrombocytopenia, anemia | [39] | |
| Wiskott–Aldrich syndrome | XL | severe | Microthrombocytopenia; Reduced α/δ granules release | [45] | |
| X-linked thrombocytopenia | XL | mild/moderate | Microthrombocytopenia; Reduced α/δ granules release | [45] | |
| Non-syndromic | ACTN1-related thrombocytopenia | AD | mild | Macrothrombocytopenia | [28] |
| Bernard Soulier syndrome monoallelic biallelic |
AD AR |
mild moderate/severe |
Macrothrombocytopenia; lack of platelet agglutination to ristocetin with normal aggregation to other agonists; severe reduction or complete lack of GPIb/IX/V | [33] | |
| CYCS-related thrombocytopenia | AD | mild | Normal platelet size and morphology | [30] | |
| FLI1-related thrombocytopenia | AD/AR | moderate | Reduced platelet aggregation in response to collagen and PAR-1 agonists; δ-granule deficiency | [15] | |
| FYB-related thrombocytopenia | AR | moderate/severe | Microthrombocytopenia; increased expression of P-selectin and PAC-1 by resting platelets but impaired upon stimulation with ADP | [16] | |
| GFI1b-related thrombocytopenia | AD/AR | mild/moderate | Macrothrombocytopenia; dyserytropoiesis; reduced α-granule content and release; diminished expression of GPIbα, red cell anisocytosis | [18] | |
| IKZF5-related thrombocytopenia | AD | absent | Thrombocytopenia; deficiency of platelet alpha granules. | [21] | |
| ITGA2B/ITGB3-related thrombocytopenia | AD | mild/moderate | Macrothrombocytopenia; reduced GPIIb/IIIa; defective GPIIb/IIIa activation (PAC-1) | [35,36,54] | |
| PT-VWD | AD | mild/moderate | Macrothrombocytopenia; increased response to ristocetin and decreased VWF-ristocetin cofactor activity (VWF:RCo) Mixing tests discriminate the plasmatic (VWD type2B) from platelet (PT-VWD) origin of hyperreactivity to ristocetin | [36,76,77] | |
| PRKACG-related thrombocytopenia | AR | severe | Macrothrombocytopenia; defective platelet αIIbβ3 activation and P-selectin exposure in response to TRAP6; defective Ca2+ mobilization in response to thrombin | [40] | |
| THPO-related thrombocytopenia | AD | mild | Normal or slightly increased platelet size | [26] | |
| TRPM7-related thrombocytopenia | AD | mild/moderate | Macrothrombocytopenia; aberrant distribution of granules | [42] | |
| Tropomyosin 4 (TPM)-related thrombocytopenia | AD | mild | Macrothrombocytopenia | [43] | |
| TUBB-1-related thrombocytopenia | AD | mild | Macrothrombocytopenia; platelet anisocytosis | [44] | |
| SLFN14-related thrombocytopenia | AD | mild/moderate | Macrothrombocytopenia; δ-granule deficiency with decreased ATP secretion in response to ADP, collagen and TRAP-6 | [49] | |
| Forms predisposing to additional diseases | ANKRD26-related thrombocytopenia | AD | mild/moderate | Reduced α-granules in some patients | [13] |
| Congenital amegakaryocytic thrombocytopenia (CAMT) | AR | severe | Elevated serum levels of TPO | [22] | |
| DIAPH1-related thrombocytopenia | AD | mild/severe | Macrothrombocytopenia | [31] | |
| ETV6-related thrombocytopenia | AD | mild/moderate | Decreased ability of platelets to spread on fibrinogen covered surfaces; abnormal clot retraction | [14] | |
| Familial platelet disorder with predisposition to hematological malignancies (FPD/AML) | AD | moderate | Abnormal aggregation in response to multiple agonists; δ (occasionally α)-granule deficiency | [25] | |
| MYH9-related disease | AD | mild/severe | Macrothrombocytopenia; Döhl-like body cytoplasmic leukocyte inclusions | [38] | |
| Thrombocytopenia associated with sitosterolemia | moderate/severe | Macrothrombocytopenia; hyperactivatable platelets with constitutive binding of fibrinogen to αIIbβ3 integrin; shedding of GPIbα; impaired platelet adhesion to von Willebrand factor | [46] |
The platelet aggregation pattern may be typical of some ITs like biallelic BSS, associated with no response to ristocetin but normal aggregation to all other agonists, or PT-VWD, with increased response to ristocetin.
Measurement of platelet glycoproteins by flow cytometry, using a well-defined set of antibodies, is the gold standard for the diagnosis of biallelic and monoallelic BSS, ITGA2B/ITGB3-RT and GFI1B-RT.
The measurement of platelet granule content and secretion can reveal alterations, e.g., in WAS and thrombocytopenia with absent radii (TAR) a reduced number of dense-granules has been reported, GPS is characterized by absent or reduced α-granules [78], Paris-Trousseau (PTS) and Jacobsen syndromes show abnormally large α-granules, while patients with FLNA-RT show some platelets having a reduced number of α-granules and others with enlarged α-granules [15,79].
Other structural abnormalities, like membranous inclusions, platelet organelle abnormalities, endoplasmic reticulum (ER)-derived inclusion bodies or particulate cytoplasmic structures with immunoreactivity for polyubiquitinated proteins and proteasome (PaCSs) [32], can be detected by electron microscopy in platelets from some specific ITs (Table 3) [18].
Additional tests may be required for complex cases, including the measurement of platelet phosphatidylserine expression, by flow cytometry, to detect enhanced procoagulant activity (Stormorken syndrome), spreading or adhesion assays to detect increased spreading in FLNA-RT, or western blotting for detection of specific proteins usually absent from platelets (e.g., MYH10 in FPD/AML and FLI1-RT).
Some additional non platelet-related laboratory tests may complement physical examination in the search for syndromic manifestations, like urinalysis, to detect proteinuria as the first sign of renal impairment in MYH9-RD, or the liver enzymes, which are elevated in approximately 50% of patients with this disease [80].
3.4. Genetic Analysis
While genotyping has mainly been used as a confirmatory test in the past, it is now playing an increasing role in the initial diagnostic approach to IT.
Until a few years ago, in fact, when the inherited nature of thrombocytopenia was suspected, a series of laboratory tests (e.g., flow cytometry for platelet surface GPs, examination of peripheral blood smear and immunofluorescence assay for MYH9 protein aggregates in neutrophils, platelet aggregometry) were performed to orient towards the candidate gene/genes to be sequenced by Sanger sequencing [66]. The application of high throughput sequencing (HTS) techniques to platelet disorders has allowed for the discovery of several novel genes associated with IT in a few years and has opened the possibility of approaching IT diagnosis by a single-step strategy. In fact, the simultaneous screening of several genes by targeted sequencing platforms, whole exome sequencing (WES) or whole genome sequencing (WGS) has been shown to provide diagnosis in 30% to 50% of patients with suspected IT [81,82,83]. Indeed, HTS is being proposed as a first line diagnostic investigation by an increasing number of authors [82,83,84,85]. However, the interpretation of genetic variants is challenging and requires a careful expert team evaluation in light of a well characterized patient phenotype [84] and when new variants in diagnostic-grade (TIER1) genes are found by targeted sequencing, WES or WGS or new genes are identified by WES or WGS it is essential that rigorous guidelines (i.e., the ACMG guidelines [86]) are applied to confirm their pathogenicity [84]. No guidelines are available yet regarding which suspected IT patients should undergo genetic testing. Some ITs with pathognonomic laboratory or clinical features, such as BSS, TAR, GATA1-RD, ATRUS, Stormorken syndrome and WAS, can be clearly diagnosed without the need of genetic testing. For other ITs, for which a strong genotype–phenotype correlation has been described, e.g., MYH9-RD, genotyping may be advisable for prognostic evaluation and possible preventive intervention. Other forms that do not have any specific diagnostic, clinical or laboratory features would require genetic testing for definite diagnosis. However, for some of these, e.g., ACTN1-RT or TUBB1-RT, a genetic diagnosis does not have any significant impact on patient management, while for others it may inform patients monitoring and treatment. Among these there are thrombocytopenias with normal platelet volume, including forms like FPD/AML, ANKRD26-RT and ETV6-RT which are predisposed to hematological malignancies (Figure 2). There are ethical implications of detecting variants in these genes and other unexpected genetic defects, such as a carrier status of a recessive gene. It is thus recommended to strictly follow an informed consent protocol ensuring that patients comprehend the possible implications of unsolicited genetic findings [85].
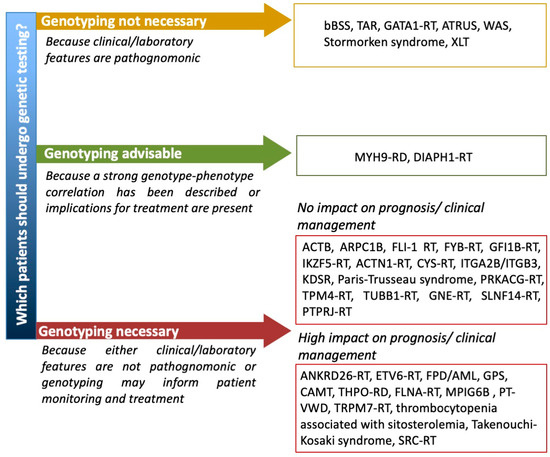
Figure 2. Proposal of a flow chart guiding the use of genetic testing for patients with suspected IT. ACTB = Baraitser–Winter syndrome 1 with macrothrombocytopenia, ARPC1B = Platelet abnormalities with eosinophilia and immune-mediated inflammatory disease, ATRUS = amegakaryocytic thrombocytopenia with radio-ulnar synostosis, bBSS = biallelic Bernard Soulier syndrome, CAMT = congenital amegakaryocytic thrombocytopenia, MPIG6B = thrombocytopenia, anemia and myelofibrosis, PT-VWD = platelet-type von Willebrand disease, RD = related disorder, RT = related thrombocytopenia, TAR = thrombocytopenia with absent radii, XLT = X-linked thrombocytopenia, WAS = Wiskott–Aldrich syndrome.
In summary, the optimal diagnostic approach to ITs is still being debated and a combination of clinical/traditional laboratory approach with advanced gene sequencing techniques may provide the highest rate of diagnostic success [69], and the best patient management.
3.5. Undefined Aspects and Possible Future Research Lines
A consensus on the classification of ITs has not been reached yet, but it would be highly advisable to avoid, for example, ambiguity on disease nomenclature.
A guidance flow chart about which suspected IT patients should undergo genetic testing is not yet available and the generation of consensus documents promoted by the relevant international scientific societies (ISTH, EHA, ASH) is highly warranted.
Moreover, development of guidelines on informed consent documents, reporting of new variants in variant databases to improve variant classification, development of user-friendly interpretation softwares of HTS results, promotion of research for discovery of new genes causing IT and development of advanced cell-based models to study platelet formation and function are valuable future perspectives.
This entry is adapted from the peer-reviewed paper 10.3390/jcm10030533
This entry is offline, you can click here to edit this entry!