Fusion RNAs are a hallmark of some cancers. They result either from chromosomal rearrangements or from splicing mechanisms that are non-chromosomal rearrangements.
- fusion RNAs
- gene fusions
- chromosomal rearrangements
- trans-splicing
1. Introduction
Cancer is a disease of the genome [1][2]. Gene fusions or chromosomal rearrangements are an important class of somatic alterations in cancer and can have important roles in the initial steps of tumorigenesis. [3][4][5]. The first cancer-associated chromosomal rearrangement was identified in 1960 as a translocation of chromosomes 9 and 22 [4][6][7]. The abnormally small resulting chromosome, named the Philadelphia chromosome, was found in over 95% of patients with chronic myelogenous leukemia (CML) and consisted of the breakpoint cluster region (BCR) gene fused to the second exon of the Abelson murine leukemia viral oncogene homolog 1 (ABL1) gene [8][9]. Additional examples of cancer-associated chromosomal aberrations have been identified in other hematological malignancies and sarcomas; for example, mixed lineage leukemia (MLL) fusions, RUNX1–RUNX1T1 and PML–RARα, EWSR1–FLI1 and EVT6–NTRK3 [10][11]. Although originally discovered in hematological malignancies, gene fusions are now known to occur in several solid tumor types [7][12][13]. The first fusion gene found in a solid tumor was CTNNB1–PLAG1 in salivary gland adenoma, which is usually benign [12][14][15]. Other fusion genes were soon discovered in solid tumors and other malignancies, including glioblastoma, melanoma, and prostate, breast, ovarian, lung, colorectal, and head and neck cancers [13].
Most of the nonprotein-coding region of the human genome was previously considered to be “junk DNA” [16]. With the advent of massive parallel sequencing technology, these regions in the human genome have been clearly shown to transcribe dynamically and differentially into noncoding RNAs (ncRNAs), such as microRNAs (miRNAs), small nucleolar RNAs (snoRNAs), long ncRNAs (lncRNAs), and circular RNAs (circRNAs) [17][18][19][20]. Accumulating evidence indicates that lncRNAs play critical roles in diverse biological processes, including differentiation, stem cell pluripotency, embryogenesis, pathogenic infection, neurogenesis, proliferation, and tumorigenesis [16][20][21][22][23][24][25][26]. LncRNAs also function in chromatin and genomic structural remodeling, RNA trafficking, RNA stabilization, transcriptional regulation, translation, signal pathway, and protein degradation [27][28][29][30][31]. Expression of lncRNA, miRNA, and snoRNA have shown close correlations with specific chromosomal rearrangements in cancers [10][32][33][34]. Moreover, fusion circRNAs (f-circRNAs) that are generated by chromosome rearrangement contribute to oncogenic roles [35]. Furthermore, the fusion RNA SLC45A3–ELK4, which regulates cancer cell proliferation, functions as a lncRNA [36]. However, details of mechanisms of the oncogenic roles of these fusion RNAs are unclear.
The ultimate goal of precision medicine in cancer treatment is the development of therapeutic strategies that specifically target cancer cells without affecting normal cells [37]. Targeting oncogenic fusion genes and RNAs specific to cancer tissue for treatment and diagnosis could bring us closer to the approach. Moreover, these fusions are often present at clonal levels within tumors; their generation is frequently the founding genetic abnormality that drives the cancer [37][38].
2. Biological Functions of Fusion Genes and RNAs
Solid tumors and hematopoietic malignancies often have highly complex, unstable genomes. Many gene fusions are random events caused by genetic instability or abnormal splicing machinery [39][40]. These changes at the gene or RNA level are unlikely to result in functional nucleic acids or proteins, as they may occur in regions where there are no known genes [12]. The functions of fusion genes and RNAs are diverse and dependent on the location of the fusion junction. However, the presence of a genomic fusion in a tumor does not necessarily mean that the fusion affects cancer development or tumorigenesis. Fusion RNAs in which the fusion junction is within the protein-coding region may be largely out-of-frame, and such out-of-frame fusions are unlikely to be functional. However, out-of-frame fusion RNAs may function as long noncoding RNAs and may exhibit regulatory functions [41].
An RNA fused between two genes in-frame is translated into a novel fusion protein that may act as a potent oncogenic driver. Kinase genes are often partners in such fusions [7][12]. Kinase fusions often retain kinase activity and result in ligand-independent constitutive activation and enhanced downstream signaling that leads to carcinogenesis [7][12]. Tyrosine kinase fusions that contain kinase-encoding genes, such as ALK, ROS1, RET, FGFR1/2/3 and NTRK1, have been detected in various types of cancer, including glioblastoma, melanoma, and carcinomas of head and neck, breast, lung, prostate, bladder, and thyroid gland [12][40][42][43]. Serine–threonine kinase fusions have also been reported [12][40][43]. These kinase fusions frequently cause activation of signaling pathways that play important roles in cell growth, survival, proliferation, and apoptosis [12][40][42][43]. In addition, kinases are ideal targets for cancer therapy; several inhibitors against kinases, such as ALK and BRAF, have been used to treat cancers with fused genes [12][44].
Fusion of transcription factors usually produces a fusion protein that leads to constitutive activation or an altered target gene, providing aberrant transcriptional machinery and cell transformation [12]. For example, the TMPRSS2–ERG fusion protein, the most common fusion in prostate cancers, mediates overexpression of E26 transformation-specific (ETS) family transcription factors in response to androgen and thus aberrantly activates downstream oncogenes that play important roles in many biological processes, including cell proliferation, angiogenesis, and invasiveness [45][46]. Moreover, the EWSR1–FLI1 fusion protein can gain the ability to bind to the genome and change the transcriptional mechanism [47][48]. Transcription factor fusions can induce a wide range of phenotypic changes that initiate or promote tumorigenesis. However, they have been generally more difficult to work with as therapeutic targets than kinase fusions [37].
3. Biosynthesis Patterns of Fusion Genes and RNAs
Fusion RNAs are known to be generated by three mechanisms [7][49][50]. The best-understood of these is chromosomal rearrangement. Two other mechanisms are grouped together as “splicing”. One is trans-splicing, in which exons from two separate RNA transcripts are spliced together. The other is cis-splicing, which involves adjacent genes on the same strand.
RNA splicing is a form of RNA processing in which a newly made precursor messenger RNA (pre-mRNA) is transformed into a mature messenger RNA (mRNA) [51][52]. It has important functions in regulating the RNA and protein diversity observed in organisms [51][52]. Pre-mRNA splicing involves recognizing and removing noncoding regions (intron excision) and the concomitant joining of coding regions (exon ligation) to produce mature mRNA. For many eukaryotic introns, splicing is performed in a series of reactions that are catalyzed by the spliceosome, a complex of small nuclear ribonucleoproteins that incorporates stepwise assembly and disassembly by several hundred proteins and five small nuclear RNAs [53][54][55]. Mechanisms for trans- and cis-splicing between neighboring genes are not well understood. In this section, we review the mechanism of fusion genes and RNA generation that are currently known.
3.1. Chromosomal Rearrangement
Gene fusions are usually caused by alterations in genomic structure resulting from DNA damage and by subsequent erroneous recombination and replication [7]. Genomic rearrangements can occur between one or two independent genes through six different known mechanisms: translocation, insertion, inversion, tandem duplication, deletion, and chromothripsis (Figure 1) [7][50][56]. Inversions, tandem duplications, and chromosomal deletions can occur within one or two adjacent genes, whereas translocations and insertions represent large-scale genomic aberrations that result from interactions between distant regions of the genome (interchromosomal rearrangements) or within the same chromosome (intrachromosomal rearrangements) [7][50][56]. As a result, gene fusions can produce aberrant fusion RNAs and proteins that may activate, reduce, or eliminate their original functions.
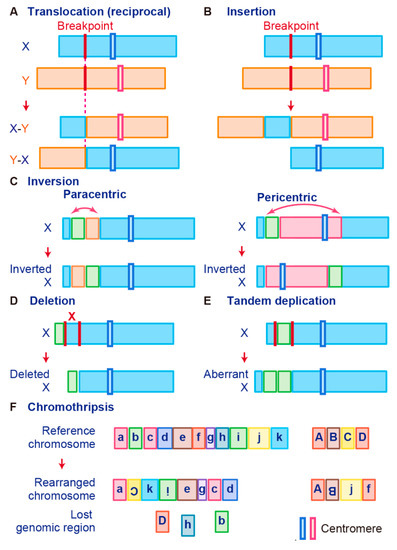
Figure 1. Schematic representation of fusion gene formation by structural chromosome rearrangements. (A) Translocation. (B) Insertion. (C) Inversion. (D) Deletion. (E) Tandem duplication. (F) Chromothripsis.
A chromosomal translocation is an exchange of parts between two nonhomologous chromosomes, also called a reciprocal translocation. Chromosomal translocation can occur anywhere between any two chromosomes (Figure 1A).
The second type of translocation is an insertion. Insertions are caused by transfer of DNA fragments from one region to another within the same chromosome (intrachromosome) or from one chromosome to another (interchromosome). The latter is also known as a nonreciprocal nonmutual translocation (Figure 1B). The BCR–ABL1, the first oncogenic fusion gene ever identified, is formed by a reciprocal chromosomal translocation. BCR–ABL1, which is generated from translocation t(9; 22) (q34; q11), is characteristic of CML [9] and also found in acute lymphoid leukemia (ALL) [57] and acute myelogenous leukemia (AML) [58]. The fusion gene BCR–ABL1 has a constitutive tyrosine kinase activity, which leads to sustained stimulation on proliferation of cancer cells [8]. One chromosomal abnormality discovered after BCR–ABL1 was a consistent nonhomologous balanced translocation between chromosomes 8 and 21 in leukemia patients [59]. This t(8; 21) translocation is one of the most common genetic defects in AML; it gives rise to the RUNX1–RUNX1T1 fusion gene (previously called AML1–ETO) [60][61][62]. The RUNX1–RUNX1T1 fusion protein interacts with other proteins to repress transcription and induce leukemogenesis in myeloid progenitor cells [60][63].
Promyelocytic leukemia (PML)–retinoic acid receptor alpha (RARα) is a fusion RNA found in almost 95% of acute promyelocytic leukemia (APL). It is generated from the t(15;17) reciprocal translocation in APL [64][65][66][67][68]. PML is a key component of PML bodies and many proteins have been associated with PML in cells [69]. The PML–RARα protein disrupts PML bodies and induces the formation of dispersed microspeckles with the loss of transcriptional activation ability [70][71]. In addition to leukemia, fusion events also occur in Ewing’s sarcoma (EWS). Many EWS cases involve t(11; 22) or t(21; 22) translocation that fuses the 5′ end of the Ewing’s sarcoma breakpoint region-1 (EWSR1) gene to the 3′ end of the FLI1 or ERG gene, which generates the fusion genes EWS–FLI1 or EWS–ERG, respectively [47][48][72]. These fusion transcription factors upregulate genes related to the cell cycle, invasion, and proliferation pathways [73][74][75][76]. Interestingly, the prion-like domain of EWS–FLI1, which is necessary for phase transitions, induces recruitment of BRG1/BRM-associated factors (BAF) complexes to GGAA microsatellites that are frequently found in oncogenes and activate the transcription of target genes [77].
A chromosome inversion occurs when a chromosome undergoes a break or rearrangement within a single chromosome (Figure 1C). There are two types of inversions: paracentric and pericentric. Paracentric inversions do not involve centromeres and both breaks occur in a single chromosome arm, whereas pericentric inversions include a centromere, with one break in each arm (Figure 1C). Many chromosomal rearrangements identified in radiation-induced tumors are known to be paracentric inversions. The most common is the RET fusion in papillary thyroid carcinoma, which is present in up to 80% of radiation-related tumors [78]. As another example, in 2007, EML4–ALK was identified as a novel fusion oncogenic driver of non-small cell lung cancer (NSCLC) [79]. The fusion of EML4 with ALK is caused by an inversion of chromosome 2 (inv2) (p21:p23), by which the kinase domain of the receptor-type tyrosine kinase ALK is placed under the control of the constitutive promoter of EML4 [80]. This fusion allows cancer transformation by activating downstream reactions in the ALK signaling pathway [80].
Chromosome deletion, which is the fourth type of rearrangement, is an alteration in which a chromosome fragment is lost during DNA replication (Figure 1D). This chromosome deletion causes the deletion of intergenic regions between two genes that are side by side, and leads to formation of fusion genes by aligning two genes that are transcribed in the same direction. An example is TMPRSS2–ERG, generated in prostate cancer via an intron deletion between TMPRSS2 and ERG on chromosome 21q22.2-3 [81][82]. Transmembrane serine protease 2 (TMPRSS2) is a prostate-specific androgen-regulated protein, and ETS-related gene (ERG) belongs to the ETS family of transcription factors, which can be oncogenic [81][82][83]. TMPRSS2–ERG fusion is reportedly associated with higher tumor stage, increased risk of disease progression, and bone metastasis [46][84].
In tandem duplication, a genomic region is duplicated and fused with a gene from the original region (Figure 1E). FGFR3–TACC3 in glioblastoma is an example of tandem duplication [85]. A tandem duplication also occurs at 7q34 in pilocytic astrocytoma, resulting in a KIAA1549–BRAF fusion gene that exhibits constitutive kinase activity. Moreover, a tandem duplication that leads to C2orf44–ALK fusion occurs in-frame on chromosome 2 in colorectal cancer, resulting in overexpression of the ALK kinase [86].
Tumor cells are associated with high genomic instability, and fusions can occur as a result of complex processes involving several rare and/or complex genetic rearrangements. Chromothripsis, which is the sixth type of rearrangement, occurs when a single chromosome, chromosome region, or a small number of chromosomes are shattered into many fragments and the fragments reassemble incorrectly (Figure 1F). Chromothripsis can produce a large number of fusion genes in a single event [87]. The most typical examples of this characteristic event are PVT1–MYC and PVT1–NDRG1 fusions in medulloblastoma. Chromothripsis in medulloblastoma leads to recurrent translocations that eventually fuse a lncRNA PVT1 to MYC, resulting in a continuous oncogenic effect via MYC amplification [88].
3.2. Trans-Splicing
In trans-splicing, exons from different RNA transcripts are spliced and fused together to produce a mature mRNA (Figure 2A) [89][90][91]. Trans-splicing produces RNAs with exon repetitions or shuffling, as well as RNAs composed of exons transcribed from opposite strands [92][93][94]. The molecular mechanisms of trans-splicing in vertebrates are largely unexplored; however, several models have been proposed [41]. One model is spliceosome-mediated trans-splicing, which uses a canonical splice site of two different primary RNAs. It shows that the spliceosome mechanism, a fundamental component of splicing, can generate trans-spliced fusion RNAs [95][96][97][98].
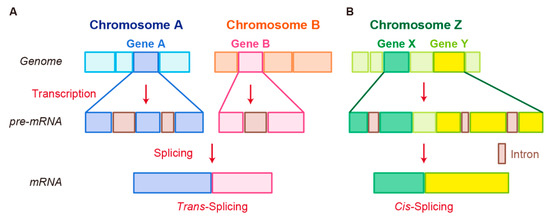
Figure 2. Schematic representation of fusion RNA formation by nonstructural chromosome rearrangements. (A) Trans-splicing. (B) Cis-splicing.
Examples of trans-spliced fusion RNAs are JAZF1–JJAZ1 (SUZ12) [89] and PAX3–FOXO1 [99]. In both cases, identical fusions were found as structural chromosomal rearrangements from human tumor tissues and RNA trans-splicing from normal human tissue. JAZF1–SUZ12, which is composed of the first 3 exons of JAZF1 and the last 15 exons of SUZ12, canonically resulted from a recurrent translocation t(7;17)(p15;q21) in endometrial stromal tumors. Identical fusion RNA was detected in normal endometrial cells [89]. Whereas PAX3–FOXO1 with t(2;13) translocation was detected in rhabdomyosarcoma, PAX3–FOXO1 fusion RNA produced by trans-splicing was transiently present in cells that underwent differentiation from pluripotent cells into skeletal muscle [99]. In these cases, different mechanisms at genomic or RNA levels may generate identical fusion RNAs, leading to different pathological outcomes.
3.3. Cis-Splicing
Another splicing mechanism is cis-splicing, in which two neighboring genes are transcribed into a single precursor RNA by transcriptional read-through, followed by RNA splicing between the exons of the two neighboring genes to complete the fusion (Figure 2B) [7][100]. Although cancer-associated fusion RNAs from cis-splicing are uncommon, some have been reported with clear oncogenic roles. SLC45A3–ELK4 has been discovered by two independent groups and is a potential biomarker in prostate cancer [101][102]. Several SLC45A3–ELK4 fusions have been reported; the primary form is a fusion of SLC45A3 exon 1 with the last four exons of ELK4. Interestingly, although SLC45A3–ELK4 functions as a fused lncRNA, its knockdown in cancer cells leads to reduction in cell proliferation, despite its unclear oncogenic roles [36]. RBM6–RBM5 is found in several types of cancer; its expression is associated with the size of breast tumors [103]. DUS4L–BCAP29 is found in gastric and prostate cancers and plays a tumor-promoting role in gastric cancer [104][105]. However, DUS4L–BCAP29 is also present in normal tissues and has a growth-promoting effect in normal as well as cancerous tissues [106].
This entry is adapted from the peer-reviewed paper 10.3390/ncrna7010010
References
- Campbell, P.J.; Getz, G.; Korbel, J.O.; Stuart, J.M.; Jennings, J.L.; Stein, L.D.; Perry, M.D.; Nahal-Bose, H.K.; Ouellette, B.F.F.; Li, C.H.; et al. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93.
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799.
- Mitelman, F.; Johansson, B.; Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 2007, 7, 233–245.
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer 2015, 15, 371–381.
- Gao, Q.; Liang, W.W.; Foltz, S.M.; Mutharasu, G.; Jayasinghe, R.G.; Cao, S.; Liao, W.W.; Reynolds, S.M.; Wyczalkowski, M.A.; Yao, L.; et al. Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep. 2018, 23, 227–238.
- Nowell, P.C.; Hungerford, D.A. Chromosome Studies on Normal and Leukemic Human Leukocytes. J. Natl. Cancer Inst. 1960, 25, 85–109.
- Dai, X.; Theobard, R.; Cheng, H.; Xing, M.; Zhang, J. Fusion genes: A promising tool combating against cancer. Biochim. Biophys. Acta-Rev. Cancer 2018, 1869, 149–160.
- Shtivelman, E.; Lifshitz, B.; Gale, R.P.; Cananni, E. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature 1985, 315, 550–554.
- Laemmli, U.K. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973, 243, 290–293.
- Han, C.; Sun, L.Y.; Wang, W.T.; Sun, Y.M.; Chen, Y.Q. Non-coding RNAs in cancers with chromosomal rearrangements: The signatures, causes, functions and implications. J. Mol. Cell Biol. 2019, 11, 886–898.
- Chen, J.; Odenike, O.; Rowley, J.D. Leukaemogenesis: More than mutant genes. Nat. Rev. Cancer 2010, 10, 23–36.
- Schram, A.M.; Chang, M.T.; Jonsson, P.; Drilon, A. Fusions in solid tumours: Diagnostic strategies, targeted therapy, and acquired resistance. Nat. Rev. Clin. Oncol. 2017, 14, 735–748.
- Parker, B.C.; Zhang, W. Fusion genes in solid tumors: An emerging target for cancer diagnosis and treatment. Chin. J. Cancer 2013, 32, 594–603.
- Kas, K.; Voz, M.L.; Roijer, E.; Astrom, A.; Meyen, E.; Stenman, G.; Ven, W.J.M. Van De Promoter swapping between the genes for a novel zinc finger protein and b-catenin in pleiomorphic adenomas with t(3;8)(p21;q12) translocations. Nat. Genet. 1997, 15, 170–174.
- Mark, J.; Dahlenfors, R.; Ekedahl, C.; Stenman, G. The mixed salivary gland tumor—A normally benign human neoplasm frequently showing specific chromosomal abnormalities. Cancer Genet. Cytogenet. 1980, 2, 231–241.
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407.
- Palazzo, A.F.; Koonin, E.V. Functional Long Non-coding RNAs Evolve from Junk Transcripts. Cell 2020, 183, 1151–1161.
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166.
- Cech, T.R.; Steitz, J.A. The noncoding RNA revolution-trashing old rules to forge new ones. Cell 2014, 157, 77–94.
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2017, 18, 5–18.
- Wong, C.M.; Tsang, F.H.; Ng, I.O.L. Non-coding RNAs in hepatocellular carcinoma: Molecular functions and pathological implications. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 137–151.
- Batista, P.J.; Chang, H.Y. Long noncoding RNAs: Cellular address codes in development and disease. Cell 2013, 152, 1298–1307.
- Huarte, M. The emerging role of lncRNAs in cancer. Nat. Med. 2015, 21, 1253–1261.
- Shirahama, S.; Miki, A.; Kaburaki, T.; Akimitsu, N. Long Non-coding RNAs Involved in Pathogenic Infection. Front. Genet. 2020, 11, 454.
- Imamura, K.; Akimitsu, N. Long non-coding RNAs involved in immune responses. Front. Immunol. 2014, 5, 3–6.
- Tano, K.; Akimitsu, N. Long non-coding RNAs in cancer progression. Front. Genet. 2012, 3, 1–6.
- Ransohoff, J.D.; Wei, Y.; Khavari, P.A. The functions and unique features of long intergenic non-coding RNA. Nat. Rev. Mol. Cell Biol. 2018, 19, 143–157.
- Taniue, K.; Kurimoto, A.; Takeda, Y.; Nagashima, T.; Okada-Hatakeyama, M.; Katou, Y.; Shirahige, K.; Akiyama, T. ASBEL-TCF3 complex is required for the tumorigenicity of colorectal cancer cells. Proc. Natl. Acad. Sci. USA 2016, 113, 12739–12744.
- Taniue, K.; Kurimoto, A.; Sugimasa, H.; Nasu, E.; Takeda, Y.; Iwasaki, K.; Nagashima, T.; Okada-Hatakeyama, M.; Oyama, M.; Kozuka-Hata, H.; et al. Long noncoding RNA UPAT promotes colon tumorigenesis by inhibiting degradation of UHRF1. Proc. Natl. Acad. Sci. USA 2016, 113, 1273–1278.
- Yanagida, S.; Taniue, K.; Sugimasa, H.; Nasu, E.; Takeda, Y.; Kobayashi, M.; Yamamoto, T.; Okamoto, A.; Akiyama, T. ASBEL, an ANA/BTG3 antisense transcript required for tumorigenicity of ovarian carcinoma. Sci. Rep. 2013, 3, 1305.
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-coding RNAs in Oncology. Cell 2019, 179, 1033–1055.
- Dixon-McIver, A.; East, P.; Mein, C.A.; Cazier, J.B.; Molloy, G.; Chaplin, T.; Lister, T.A.; Young, B.D.; Debernardi, S. Distinctive patterns of microRNA expression associated with karyotype in acute myeloid leukaemia. PLoS ONE 2008, 3, e2141.
- Garzon, R.; Volinia, S.; Liu, C.G.; Fernandez-Cymering, C.; Palumbo, T.; Pichiorri, F.; Fabbri, M.; Coombes, K.; Alder, H.; Nakamura, T.; et al. MicroRNA signatures associated with cytogenetics and prognosis in acute myeloid leukemia. Blood 2008, 111, 3183–3189.
- Li, Z.; Lu, J.; Sun, M.; Mi, S.; Zhang, H.; Luo, R.T.; Chen, P.; Wang, Y.; Yan, M.; Qian, Z.; et al. Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc. Natl. Acad. Sci. USA 2008, 105, 15535–15540.
- Guarnerio, J.; Bezzi, M.; Jeong, J.C.; Paffenholz, S.V.; Berry, K.; Naldini, M.M.; Lo-Coco, F.; Tay, Y.; Beck, A.H.; Pandolfi, P.P. Oncogenic Role of Fusion-circRNAs Derived from Cancer-Associated Chromosomal Translocations. Cell 2016, 165, 289–302.
- Qin, F.; Zhang, Y.; Liu, J.; Li, H. SLC45A3-ELK4 functions as a long non-coding chimeric RNA. Cancer Lett. 2017, 404, 53–61.
- Brien, G.L.; Stegmaier, K.; Armstrong, S.A. Targeting chromatin complexes in fusion protein-driven malignancies. Nat. Rev. Cancer 2019, 19, 255–269.
- Crompton, B.D.; Stewart, C.; Taylor-Weiner, A.; Alexe, G.; Kurek, K.C.; Calicchio, M.L.; Kiezun, A.; Carter, S.L.; Shukla, S.A.; Mehta, S.S.; et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014, 4, 1326–1341.
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 340, 1546–1558.
- Yoshihara, K.; Wang, Q.; Torres-Garcia, W.; Zheng, S.; Vegesna, R.; Kim, H.; Verhaak, R.G.W. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene 2015, 34, 4845–4854.
- Chwalenia, K.; Facemire, L.; Li, H. Chimeric RNAs in cancer and normal physiology. Wiley Interdiscip. Rev. RNA 2017, 8.
- Kim, H.J.; Kim, N.C.; Wang, Y.D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473.
- Stransky, N.; Cerami, E.; Schalm, S.; Kim, J.L.; Lengauer, C. The landscape of kinase fusions in cancer. Nat. Commun. 2014, 5, 4846.
- Shaw, A.T.; Hsu, P.P.; Awad, M.M.; Engelman, J.A. Tyrosine kinase gene rearrangements in epithelial malignancies. Nat. Rev. Cancer 2013, 13, 772–787.
- Rabbitts, T.H. Commonality but Diversity in Cancer Gene Fusions. Cell 2009, 137, 391–395.
- Deplus, R.; Delliaux, C.; Marchand, N.; Flourens, A.; Vanpouille, N.; Leroy, X.; de Launoit, Y.; Duterque-Coquillaud, M. TMPRSS2-ERG fusion promotes prostate cancer metastases in bone. Oncotarget 2017, 8, 11827–11840.
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; De Jong, P.; Rouleau, G.; et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992, 359, 162–165.
- May, W.A.; Gishizky, M.L.; Lessnick, S.L.; Lunsford, L.B.; Lewis, B.C.; Delattre, O.; Zucman, J.; Thomas, G.; Denny, C.T. Ewing sarcoma 11;22 translocation produces a chimeric transcription factor that requires the DNA-binding domain encoded by FLI1 for transformation. Proc. Natl. Acad. Sci. USA 1993, 90, 5752–5756.
- Li, Z.; Qin, F.; Li, H. Chimeric RNAs and their implications in cancer. Curr. Opin. Genet. Dev. 2018, 48, 36–43.
- Tuna, M.; Amos, C.I.; Mills, G.B. Molecular mechanisms and pathobiology of oncogenic fusion transcripts in epithelial tumors. Oncotarget 2019, 10, 2095–2111.
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463.
- Irimia, M.; Blencowe, B.J. Alternative splicing: Decoding an expansive regulatory layer. Curr. Opin. Cell Biol. 2012, 24, 323–332.
- Jurica, M.S.; Moore, M.J. Pre-mRNA splicing: Awash in a sea of proteins. Mol. Cell 2003, 12, 5–14.
- Ule, J.; Blencowe, B.J. Alternative Splicing Regulatory Networks: Functions, Mechanisms, and Evolution. Mol. Cell 2019, 76, 329–345.
- Shi, Y. Mechanistic insights into precursor messenger RNA splicing by the spliceosome. Nat. Rev. Mol. Cell Biol. 2017, 18, 655–670.
- Neckles, C.; Sundara Rajan, S.; Caplen, N.J. Fusion transcripts: Unexploited vulnerabilities in cancer? Wiley Interdiscip. Rev. RNA 2020, 11, 1–17.
- Lucas, G.S.; Ardern, J.C.; Bartram, C.R. BCR-ABL rearrangements in acute lymphoblastic leukaemia. Lancet 1991, 337, 1548.
- Cuenco, G.M.; Ren, R. Cooperation of BCR-ABL and AML1/MDS1/EVI1 in blocking myeloid differentiation and rapid induction of an acute myelogenous leukemia. Oncogene 2001, 20, 8236–8248.
- Nucifora, G.; Birn, D.J.; Erickson, P.; Gao, J.; LeBeau, M.M.; Drabkin, H.A.; Rowle, J.D. Detection of DNA rearrangements in the AML1 and ETO loci and of an AML1/ETO fusion mRNA in patients with t(8;21) acute myeloid leukemia. Blood 1993, 81, 883–888.
- Higuchi, M.; O’Brien, D.; Kumaravelu, P.; Lenny, N.; Yeoh, E.J.; Downing, J.R. Expression of a conditional AML1-ETO oncogene bypasses embryonic lethality and establishes a murine model of human t(8;21) acute myeloid leukemia. Cancer Cell 2002, 1, 63–74.
- Jurlander, J.; Caligiuri, M.A.; Ruutu, T.; Baer, M.R.; Strout, M.P.; Oberkircher, A.R.; Hoffmann, L.; Ball, E.D.; Frei-Lahr, D.A.; Christiansen, N.P.; et al. Persistence of the AML1/ETO fusion transcript in patients treated with allogeneic bone marrow transplantation for t(8;21) leukemia. Blood 1996, 88, 2183–2191.
- Nucifora, G.; Rowley, J.D. AML1 and the 8;21 and 3;21 translocations in acute and chronic myeloid leukemia. Blood 1995, 86, 1–14.
- Yuan, Y.; Zhou, L.; Miyamoto, T.; Iwasaki, H.; Harakawa, N.; Hetherington, C.J.; Burel, S.A.; Lagasse, E.; Weissman, I.L.; Akashi, K.; et al. AML1-ETO expression is directly involved in the development of acute myeloid leukemia in the presence of additional mutations. Proc. Natl. Acad. Sci. USA 2001, 98, 10398–10403.
- de Thé, H.; Lavau, C.; Marchio, A.; Chomienne, C.; Degos, L.; Dejean, A. The PML-RARα fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell 1991, 66, 675–684.
- Goddard, A.D.; Borrow, J.; Freemont, P.S.; Solomon, E. Characterization of a zinc finger gene disrupted by the t(15;17) in acute promyelocytic leukemia. Science 1991, 254, 1371–1374.
- Kakizuka, A.; Miller, W.H.; Umesono, K.; Warrell, R.P.; Frankel, S.R.; Murty, V.V.V.S.; Dmitrovsky, E.; Evans, R.M. Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RARα with a novel putative transcription factor, PML. Cell 1991, 66, 663–674.
- Meroni, G.; Diez-Roux, G. TRIM/RBCC, a novel class of “single protein RING finger” E3 ubiquitin ligases. BioEssays 2005, 27, 1147–1157.
- Reymond, A.; Meroni, G.; Fantozzi, A.; Merla, G.; Cairo, S.; Luzi, L.; Riganelli, D.; Zanaria, E.; Messali, S.; Cainarca, S.; et al. The tripartite motif family identifies cell compartments. EMBO J. 2001, 20, 2140–2151.
- Van Damme, E.; Laukens, K.; Dang, T.H.; Van Ostade, X. A manually curated network of the PML nuclear body interactome reveals an important role for PML-NBs in SUMOylation dynamics. Int. J. Biol. Sci. 2010, 6, 51–67.
- Shima, Y.; Shima, T.; Chiba, T.; Irimura, T.; Pandolfi, P.P.; Kitabayashi, I. PML Activates Transcription by Protecting HIPK2 and p300 from SCFFbx3-Mediated Degradation. Mol. Cell. Biol. 2008, 28, 7126–7138.
- Di Masi, A.; Cilli, D.; Berardinelli, F.; Talarico, A.; Pallavicini, I.; Pennisi, R.; Leone, S.; Antoccia, A.; Noguera, N.I.; Lo-Coco, F.; et al. PML nuclear body disruption impairs DNA double-strand break sensing and repair in APL. Cell Death Dis. 2016, 7, e2308.
- Sorensen, P.H.B.; Lessnick, S.L.; Lopez-terrada, D.; Liu, X.F.; Triche, T.J.; Denny, C.T.; Cherry, H. A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS–family transcription factor, ERG. Nat. Genet. 1994, 6, 146–151.
- Zinszner, H.; Immanuel, D.; Yin, Y.; Liang, F.X.; Ron, D. A topogenic role for the oncogenic N-terminus of TLS: Nucleolar localization when transcription is inhibited. Oncogene 1997, 14, 451–461.
- Yang, L.; Chansky, H.A.; Hickstein, D.D. EWS·Fli-1 fusion protein interacts with hyperphosphorylated RNA polymerase II and interferes with serine-arginine protein-mediated RNA splicing. J. Biol. Chem. 2000, 275, 37612–37618.
- Bailly, R.; Bosselut, R.; Zucman, J.; Delattre, O.; Roussel, M.; Thomas, G.; Ghysdaell, J. DNA-binding and transcriptional activation properties of the EWS-FLI-1 fusion protein resulting from the t (11; 22) translocation in Ewing sarcoma. Mol. Cell. Biol. 1994, 14, 3230–3241.
- Braun, B.S.; Frieden, R.; Lessnick, S.L.; May, W.A.; Denny, C.T. Identification of target genes for the Ewing’s sarcoma EWS/FLI fusion protein by representational difference analysis. Mol. Cell. Biol. 1995, 15, 4623–4630.
- Boulay, G.; Sandoval, G.J.; Riggi, N.; Iyer, S.; Buisson, R.; Naigles, B.; Awad, M.E.; Rengarajan, S.; Volorio, A.; McBride, M.J.; et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 2017, 171, 163–178.
- Pierotti, M.A.; Santoro, M.; Jenkins, R.B.; Sozzi, G.; Bongarzone, I.; Grieco, M.; Monzini, N.; Miozzo, M.; Herrmann, M.A.; Fusco, A.; et al. Characterization of an inversion on the long arm of chromosome 10 juxtaposing D10S170 and RET and creating the oncogenic sequence RET/PTC. Proc. Natl. Acad. Sci. USA 1992, 89, 1616–1620.
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.I.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566.
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global Survey of Phosphotyrosine Signaling Identifies Oncogenic Kinases in Lung Cancer. Cell 2007, 131, 1190–1203.
- Pederzoli, F.; Bandini, M.; Marandino, L.; Ali, S.M.; Madison, R.; Chung, J.; Ross, J.S.; Necchi, A. Targetable gene fusions and aberrations in genitourinary oncology. Nat. Rev. Urol. 2020, 17, 613–625.
- Tomlins, S.A.; Laxman, B.; Dhanasekaran, S.M.; Helgeson, B.E.; Cao, X.; Morris, D.S.; Menon, A.; Jing, X.; Cao, Q.; Han, B.; et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 2007, 448, 595–599.
- Hollenhorst, P.C.; McIntosh, L.P.; Graves, B.J. Genomic and biochemical insights into the specificity of ETS transcription factors. Annu. Rev. Biochem. 2011, 80, 437–471.
- Perner, S.; Demichelis, F.; Beroukhim, R.; Schmidt, F.H.; Mosquera, J.M.; Setlur, S.; Tchinda, J.; Tomlins, S.A.; Hofer, M.D.; Pienta, K.G.; et al. TMPRSS2:ERG fusion-associated deletions provide insight into the heterogeneity of prostate cancer. Cancer Res. 2006, 66, 8337–8341.
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Porrati, P.; Pellegatta, S.; et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012, 337, 1231–1235.
- Lipson, D.; Capelletti, M.; Yelensky, R.; Otto, G.; Parker, A.; Jarosz, M.; Curran, J.A.; Balasubramanian, S.; Bloom, T.; Brennan, K.W.; et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat. Med. 2012, 18, 382–384.
- Holland, A.J.; Cleveland, D.W. Chromoanagenesis and cancer: Mechanisms and consequences of localized, complex chromosomal rearrangements. Nat. Med. 2012, 18, 1630–1638.
- Northcott, P.A.; Shih, D.J.H.; Peacock, J.; Garzia, L.; Sorana Morrissy, A.; Zichner, T.; Stútz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 2012, 487, 49–56.
- Li, H.; Wang, J.; Mor, G.; Sklar, J. A Neoplastic Gene Fusion Mimics Trans-Splicing of RNAs in Normal Human Cells. Science 2008, 321, 1357–1361.
- Li, B.L.; Li, X.L.; Duan, Z.J.; Lee, O.; Lin, S.; Ma, Z.M.; Chang, C.C.Y.; Yang, X.Y.; Park, J.P.; Mohandas, T.K.; et al. Human acyl-CoA:cholesterol acyltransferase-1 (ACAT-1) gene organization and evidence that the 4.3-kilobase ACAT-1 mRNA is produced from two different chromosomes. J. Biol. Chem. 1999, 274, 11060–11071.
- Breen, M.A.; Ashcroft, S.J.H. A truncated isoform of Ca2+/calmodulin-dependent protein kinase II expressed in human islets of Langerhans may result from trans-splicing. FEBS Lett. 1997, 409, 375–379.
- Horiuchi, T.; Giniger, E.; Aigaki, T. Alternative trans-splicing of constant and variable exons of a Drosophila axon guidance gene, lola. Genes Dev. 2003, 17, 2496–2501.
- Dorn, R.; Reuter, G.; Loewendorf, A. Transgene analysis proves mRNA trans-splicing at the complex mod(mdg4) locus in Drosophila. Proc. Natl. Acad. Sci. USA 2001, 98, 9724–9729.
- Lei, Q.; Li, C.; Zuo, Z.; Huang, C.; Cheng, H.; Zhou, R. Evolutionary Insights into RNA trans-Splicing in Vertebrates. Genome Biol. Evol. 2016, 8, 562–577.
- Solnick, D. Trans splicing of mRNA precursors. Cell 1985, 42, 157–164.
- Konarska, M.M.; Padgett, R.A.; Sharp, P.A. Trans splicing of mRNA precursors in vitro. Cell 1985, 42, 165–171.
- Eul, J.; Graessmann, M.; Graessmann, A. Experimental evidence for RNA trans-splicing in mammalian cells. EMBO J. 1995, 14, 3226–3235.
- Eul, J.; Graessmann, M.; Graessmann, A. Trans-splicing and alternative-tandem-cis-splicing: Two ways by which mammalian cells generate a truncated SV40 T-antigen. Nucleic Acids Res. 1996, 24, 1653–1661.
- Yuan, H.; Qin, F.; Movassagh, M.; Park, H.; Golden, W.; Xie, Z.; Zhang, P.; Sklar, J.; Li, H. A chimeric RNA characteristic of rhabdomyosarcoma in normal myogenesis process. Cancer Discov. 2013, 3, 1394–1403.
- Wu, H.; Li, X.; Li, H. Gene fusions and chimeric RNAs, and their implications in cancer. Genes Dis. 2019, 6, 385–390.
- Maher, C.A.; Kumar-Sinha, C.; Cao, X.; Kalyana-Sundaram, S.; Han, B.; Jing, X.; Sam, L.; Barrette, T.; Palanisamy, N.; Chinnaiyan, A.M. Transcriptome sequencing to detect gene fusions in cancer. Nature 2009, 458, 97–101.
- Rickman, D.S.; Pflueger, D.; Moss, B.; VanDoren, V.E.; Chen, C.X.; De la Taille, A.; Kuefer, R.; Tewari, A.K.; Setlur, S.R.; Demichelis, F.; et al. SLC45A3-ELK4 is a novel and frequent erythroblast transformation-specific fusion transcript in prostate cancer. Cancer Res. 2009, 69, 2737–2738.
- Wang, K.; Ubriaco, G.; Sutherland, L.C. RBM6-RBM5 transcription-induced chimeras are differentially expressed in tumours. BMC Genom. 2007, 8, 1–12.
- Nacu, S.; Yuan, W.; Kan, Z.; Bhatt, D.; Rivers, C.S.; Stinson, J.; Peters, B.A.; Modrusan, Z.; Jung, K.; Seshagiri, S.; et al. Deep RNA sequencing analysis of readthrough gene fusions in human prostate adenocarcinoma and reference samples. BMC Med. Genom. 2011, 4, 11.
- Kim, H.P.; Cho, G.A.; Han, S.W.; Shin, J.Y.; Jeong, E.G.; Song, S.H.; Lee, W.C.; Lee, K.H.; Bang, D.; Seo, J.S.; et al. Novel fusion transcripts in human gastric cancer revealed by transcriptome analysis. Oncogene 2014, 33, 5434–5441.
- Tang, Y.; Qin, F.; Liu, A.; Li, H. Recurrent fusion RNA DUS4L-BCAP29 in non-cancer human tissues and cells. Oncotarget 2017, 8, 31415–31423.