Atherosclerosis is recognized as a chronic inflammatory disease characterized by the accumulation of lipids, mainly cholesterol, and other components, such as fatty substances, cellular waste products, calcium, and fibrin within the arterial wall.
- atherosclerosis
- mitochondria
- inflammation
- inflammasome
- reactive oxygen species
- NLRP3
1. Mitochondria and Inflammation
Mitochondria are one of the most multifunctional organelles in the cell [1]. Their major function as cell energy generators has been extensively studied. In addition, mitochondria are involved in many cell processes, such as steroid biosynthesis [2], calcium [3] and iron [4] homeostasis, immune cell activation [5], redox signaling [6], apoptosis [7], and inflammation [8].
The first line of defense of metazoan organisms to deal with infection and/or tissue damage is the innate immunity response [9]. During infection, inflammation is commonly caused by microbial compounds, known as pathogen-associated molecule patterns (PAMPs), for instance, lipopolysaccharides (LPS) from bacteria or viral RNA. On the other hand, during tissue damage, inflammation is activated by intracellular molecules that are not usually exposed to the immune system. However, during cell stress or damage, they are secreted into the cytoplasm or leak into the extracellular environment. These molecules are called damage-associated molecular patterns (DAMPs).
Pattern recognition receptors (PRRs), including Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), NOD-like receptors (NLRs), and C-type lectin receptors (CLRs), are sensors that recognize both PAMPs and DAMPs. The ligation of PRRs by DAMPs induces intracellular signaling pathways that promote the expression and activation of several pro-inflammatory mechanisms whose regulation and response depend on the PRR and cell type. Curiously, some PRR can ligate with both DAMPs and PAMPs [10]. The stimulation of a PRR through its ligands triggers the activation of the immune system [11]. For instance, DAMPs such as uric acid promote dendritic cell maturation [12] while PAMPs such as chitin, a major component of the fungal cell wall, can be recognized by epidermal cells for chemokine secretion and leukocyte recruitment [13]. Normally, the sensing of PAMPs or DAMPs by PRRs leads to the nuclear translocation of transcription factors, including the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which upregulate the transcription of genes involved in the expression of pro-inflammatory cytokines, type I interferons (IFNs), chemokines and antimicrobial proteins, proteins involved in the modulation of PRR signaling, and inflammasome proteins [14].
The close similarity between prokaryotic DNA and mitochondrial DNA (mtDNA) is an important factor for understanding the role of mitochondria in inflammation, as well as supporting evidence for the endosymbiosis theory [15]. mtDNA is a double-stranded, circular molecule that contains a high concentration of cytosine-guanine sequences or CpG islands. mtDNA is released by damaged cells and can be sensed by a PRR, the Toll-like receptor 9 (TLR9), which is the receptor for CpG motifs in DNA [16]. This interaction leads to the NF-κB activation signaling pathway and, consequently, the induction of multiple pro-inflammatory genes [17][18]. In addition, mtDNA can activate the nod like receptor family pyrin domain containing 3 (NLRP3) inflammasome [19], thereafter promoting caspase-1 activation, the processing of interleukin 1β (IL-1β) and interleukin 18 (IL-18), and eventually, cell death. Mitochondrial dysfunction may also amplify the activation of NLRP3 by mitochondrial ROS production. It is well known that these oxygen species bind and activate NLRP3 in a continuous vicious cycle where NLRP3 will also promote ROS generation [20]. The stimulator of interferon genes (STING) inflammatory pathway can also be activated via mtDNA by the protein cyclic GMP-AMP synthase (cGAS) [21]. In this case, it will result in increased interferon-regulatory factor 3 (IRF3)-dependent gene expression, including induction of type I interferons. In summary, mtDNA is able to activate the innate immune signaling pathways by acting as a danger signal released from mitochondria into the cytoplasm, which warns the cell of serious damage.
Mitochondria are also necessary for the oligomerization of retinoic-acid-inducible gene I (RIG-I)-like receptors (RLR), which are involved in the recognition of the viral double-stranded RNA PAMP. After viral RNA is detected by RIG-I in the cytosol, it interacts with the mitochondrial antiviral signaling protein (MAVS), located in the outer mitochondrial membrane. Then MAVS is activated and recruits the machinery involved in type I interferon production [8][22]. MAVS also participates in other signal pathways related with mitochondrial reactive oxygen species (mtROS) and inflammation [23][24]. Mitochondrial activity is also necessary for the Toll-like receptor (TLR) signaling cascades: activated TLRs can signal through tumor-necrosis-factor-receptor-associated factor 6 (TRAF), which translocates to the mitochondrion and ubiquitinates the mitochondrial protein evolutionarily conserved signaling intermediate in Toll pathway (ECSIT). This causes the mitochondrion to both move to the phagosome and enhance mtROS production, resulting in direct antimicrobial killing [25][26]. For instance, TLR1, TLR2, and TLR4 recognize bacterial tri-acylated lipopeptides and LPS in order to enhance ROS production in the phagosome (oxidative burst of macrophages), which is required for its antimicrobial activity [27].
2. Atherosclerosis as a Representative Inflammatory Disease
Atherosclerosis is recognized as a chronic inflammatory disease characterized by the accumulation of lipids, mainly cholesterol, and other components, such as fatty substances, cellular waste products, calcium, and fibrin within the arterial wall [28]. Atherosclerosis is virtually the primary cause of cardiovascular disease (CVD)-related events, including myocardial infarction and stroke. CVD is the leading cause of mortality worldwide, accounting for almost 17 million deaths every year [29], approximately one-third of the total global deaths. The development of atherosclerosis is primordially thought to be the result of a previous dyslipidemia, mostly hyperlipidemia [30]. However, the precise initiating event of atherosclerosis seems to be multifactorial and remains unknown. Both high-density lipoprotein (HDL) and low-density lipoprotein (LDL) are essential for cholesterol transport and have been associated with atherosclerosis progression [31]. Specifically, elevated levels of LDL cholesterol (LDL-C) have been related to atherosclerosis progression [32]. LDLs retained in the extracellular matrix mainly by proteoglycans become targets for oxidative and enzymatic modifications. Then, oxidized LDLs (oxLDLs) lead to pro-inflammatory reactions, promoting the activation and recruitment of monocytes and other inflammatory cells trafficking across the vessel wall [33]. Mutations in proteins related with cholesterol production and transport, such as LDL receptor (LDL-R) and apolipoprotein B (Apo B), will usually result in familial hypercholesterolemia, a group of genetic disorders characterized by highly elevated plasma total-cholesterol levels and an early onset of atherosclerosis [34][35].
Several studies support not only lipid accumulation but also the key role of the inflammatory mechanisms in atherosclerosis progression [36][37][38]. To sum up, the pathomechanisms of atherosclerosis involve the following steps: (1) Endothelial cells suffer inflammatory activation caused by oxLDL [39]. (2) Monocytes and other leukocytes are recruited in response to endothelium stress. (3) Chemokines and chemoattractant proteins alert inflammatory cells to migrate into the intima layer. (4) Monocytes differentiate into macrophages, which internalize LDL-C and transform into foam cells. (5) Foam cells and inflammatory macrophages release inflammatory cytokines and ROS. (6) Accumulation of dead foam cells will eventually form the lipid or necrotic core of the mature plaque. (7) The arrival of new immune cells amplifies the local inflammatory response and weakens the plaque’s fibrous cap. In addition, matrix metalloproteases (MMPs) promote vascular smooth muscle growth, while conversely provoking focal destruction of the vascular extracellular matrix, facilitating plaque rupture [40]. (8) Finally, the fracture or erosion of a weakened fibrous cap allows the exposition of thrombogenic components of the necrotic core of the plaque [41]. These steps are represented in Figure 1. As mentioned above, inflammation has a relevant role in the first steps of atherosclerotic lesions and promotes most of the thrombotic plaque complications [42].
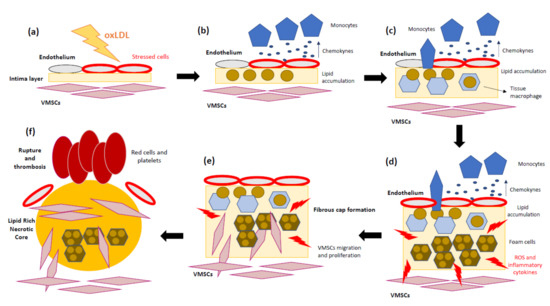
Figure 1. Schematic overview of atherosclerosis progression. (a) Oxidized low-density lipoprotein (oxLDL) contributes to the initial lesion in the arterial wall. (b) Endothelium cells produce pro-inflammatory cytokines, and circulating monocytes are recruited. (c) Monocytes migrate into the intima and differentiate into tissue macrophages. (d) Once in the artery wall, macrophages engulf the excessive lipids and become lipid-laden foam cells, which can accumulate and form a fatty streak. During the complex lesion progression, foam cell lysis by apoptosis and necrosis leads to the formation of a necrotic core and, together with defective efferocytosis, leads to the amplification of the inflammatory response. (e) Vascular smooth muscle cells (VSMCs) migrate from the media to the intima, where they differentiate into proliferative synthetic cells that generate extracellular matrix to form the fibrous cap and hence stabilize plaques. (f) During later stages, the plaque can become unstable due to the inhibition of extracellular matrix (ECM) formation, particularly collagen production by VSMCs. In addition, ECM is degraded by proteases released by macrophages, resulting in an unstable lesion that can rupture and lead to thrombosis.
Inflammatory responses can be triggered by the formation of the inflammasome, a cytoplasmic multimeric protein complex that is assembled in response to DAMPs or PAMPs [43]. The canonical inflammasome assembly starts with the activation of nod-like receptors (NLRs) or AIM2-like receptors (ALR) that induce the initial inflammatory signaling. The inflammasome holds the NLR and ALR sensors, adaptors such as Apoptosis-associated Speck-like protein containing a Caspase recruitment domain (ASC), and effectors such as caspase-1 [44]. Caspase-1 is considered an interleukin 1 (IL-1) converting enzyme (ICE) [45]. Through inflammasome activation, caspase-1 is able to convert pro-IL-1β into mature IL-1β, which is secreted to initiate inflammatory responses. Caspase-1 can also maturate pro-IL-18 into IL-18, which is related with IL-1 family cytokines [46]. Several distinct inflammasomes have been identified, among them some named after the NLR protein they contain, such as NLRP1, NLRP3, NLRC4, NLRP6, and NLRP12 [47]. Thus, for instance, NLRP3 inflammasomes are composed of NLRP3 protein, ASC, and caspase-1 [48]. Inflammasomes can be activated by diverse factors from different sources, such as extracellular adenosine triphosphate (ATP) or intracellular mtDNA. However, it is still unknown how NLRP3 can detect DAMPs [49]. There are very clear NLRP3 inflammasome activators, such as alterations in the potassium (K+) efflux, generation of mitochondrial ROS, and lysosomal destabilization [50]. Two main signals are necessary for NLRP3 inflammasome activation: The first one is called priming and requires signaling through pattern recognition receptors, such as TLRs, and the subsequent activation of NF-kB, which triggers the transcription of NLRP3 protein. The second one requires another activating signal, either mtDNA or ROS. Subsequently, NLRP3 assembles with ASC, and this complex recruits pro-caspase-1 for its maturation into its active form. Once the NLRP3 inflammasome is fully formed, pro-IL-β is processed into mature IL-1β, which is released by a mechanism independent of the Golgi apparatus [51][52].
As several experimental and clinical reports have demonstrated that IL-1β is a proatherogenic cytokine [53][54], NLRP3 inflammasome activation could contribute to the progression of atherosclerosis. Duewell et al. [55] demonstrated the relationship between NLRP3 inflammasomes and atherosclerosis. Using atherosclerosis-prone LDLR−/− mice, they produced chimeric mice whose bone marrows were transplanted with NLRP3−/−, ASC−/−, or IL-1α/β−/− bone marrow cells and showed that the lack of these inflammasome-related molecules significantly reduced the development of atherosclerotic lesions. Because cholesterol crystal formation is present in the early stages and can be detected in all stages of atherosclerosis, Duewell et al. focused on cholesterol crystals as a DAMP candidate and found that these crystals strongly activated NLRP3 inflammasomes in macrophages. Previously, it had been demonstrated that the presence of oxLDL can lead to cholesterol crystallization [56]. Therefore, the induction of NLRP3 and pro-IL-1β expression by oxLDL could promote IL-1β release. In addition, incorporation of oxLDL via a scavenger receptor CD36 provoked intracellular cholesterol crystallization in endothelial cells [57][58].
Nowadays, the NLRP3 inflammasome, a main generator of activated IL-1 family cytokines, is extensively studied due to its crucial role in the pathogenesis of atherosclerosis. Although NLRP3 inflammasome regulation is not clear, some mechanisms have been elucidated, such as its ubiquitination and phosphorylation, which induce post-transcriptional modifications that modulate its activation [59]. Until now, just one report has shown that hematopoietic NLRP3 deficiency in an LDLR−/−mouse model has small influences on atherogenesis progression [60]. In contrast, numerous studies have manifested the relevant significance of the NLRP3 inflammasome in atherosclerosis. For example, separate studies have verified that ApoE−/− Caspase-1−/− mice on an atherogenic diet show a significant reduction in atherosclerotic lesions [61][62]. In another study, NLRP3 downregulation in a diet-induced model of atherogenesis in double ApoE−/−mice prevented plaque progression and inhibited the maturation of pro-inflammatory cytokines [63].
Ultimately, NLRP3 activation in endothelial cells triggers the release of IL-1β [64]. Thus, increased levels of IL-1β were observed in atherosclerotic patients and were positively correlated to disease severity [65]. Furthermore, IL-1β acts on most cells present in the atheroma. In the endothelium, it induces procoagulant activity, increased expression of adhesion molecules for leukocyte recruitment, and production of monocyte chemoattractant protein 1 (MCP-1) [66]. All these changes allow the recruitment of monocytic phagocytes, which are strongly implicated in atherogenesis. IL-1β also acts on human vascular smooth muscle cells (VSMCs), a cell type that participates in all stages of atherosclerotic plaques, promoting their proliferation [67]. In this respect, it is worth mentioning the Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS) trial [58], a massive a randomized, double-blinded, placebo-controlled trial that investigated the use of canakinumab, a monoclonal antibody targeting IL-1β, on high-risk patients with established atherosclerotic disease who had already survived myocardial infarction. It was concluded that anti-inflammatory therapy targeting the IL-1β innate immunity pathway with canakinumab led to a significantly lower rate of recurrent cardiovascular events independent of lipid-level lowering, therefore bringing out the importance of inflammation in atherosclerosis [68].
There is a clear correlation between aortic NLRP3 expression and CVD prevalence [69]. It has been found that patients with coronary atherosclerosis display high aortic expression of NLRP3, which correlates with its severity [70]. Moreover, there is a significantly up-regulation of NLRP3 inflammasome components as well as mature IL-1β and IL-18 in human carotid atherosclerotic plaque tissue obtained by carotid endarterectomy in comparison to non-atherosclerotic mesenteric or iliac arteries [71][72]. NLRP3 expression levels were significantly higher in symptomatic compared with asymptomatic patients [72], and NLRP3 inflammasome components and signaling were highly expressed in unstable compared with stable atherosclerotic plaques [71]. For this reason, it could be interesting to consider NLRP3 protein expression levels in peripheral blood monocytes as a biomarker for predicting atherogenic complications in patients with CVD [73].
3. The Role of Mitochondria in Atherosclerosis
Mitochondria are the major source of energy in the cell through their mitochondrial respiratory chain (MRC). Almost all the energy released during mitochondrial electron transport is used for ATP synthesis, making MRC one of the most refined processes in nature. However, just a small percentage of electrons leak to oxygen, resulting in the generation of superoxide radicals, which are considered mitochondrial ROS [74]. There are alternative sources of hydrogen peroxide (H2O2) and superoxide radical anion (O2−), such as the mitochondrial outer membrane enzymes monoamine oxidase [75] and aldehyde oxidase [76].
ROS are necessary for the cell as secondary signaling agents [77], and they are regulated by numerous antioxidant molecules and proteins. In fact, ROS are gaining relevance in many research fields since it is now known that they can function as signaling molecules and in protein modification processes [78][79]. This mechanism is carried out by the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzymes, whose sole function is the formation of ROS. Seven NADPH oxidases are expressed in the human body, namely Nox1–Nox5 and Duox1 and Duox2 [80]. Each one has different functions, for instance, Nox2 is involved in host defense [79], Nox4 plays a role in cellular homeostasis and cancer [81], and Nox5 is related with cardiovascular health [82].
In mitochondria, O2- is rapidly transformed into H2O2 by manganese-dependent superoxide dismutase (SOD2), followed by its conversion into water by glutathione peroxidase 1 (GPX1) [83]. Under stress or pathological conditions, ROS are released from different sources, such as the activity of xanthine oxidase, lipoxygenase, nicotinamide adenine dinucleotide phosphate oxidase, uncoupling of nitric oxide (NO) synthase, and the leakage of electrons at mitochondrial complexes I and III during oxidative phosphorylation (OXPHOS) [84]. Therefore, damaged mitochondria produce large amounts of ROS, which in turn can affect the function of adjacent mitochondria. This continuous oxidative cycle is named ROS-induced ROS and is a common phenomenon based on the amplification of ROS production that induces further mitochondrial and cell dysfunction [85]. Interestingly, it has been reported that cytochrome c, a heme-containing protein mainly involved in mitochondrial electron transport, is involved in the ROS-induced ROS process [86].
Under normal physiological conditions, ROS damage is controlled by antioxidant molecules, such as glutathione, carotenoids, coenzyme Q10 (CoQ10), and antioxidant enzymes. However, in atherosclerosis, when ROS surpass the antioxidant barriers, the increased oxidative stress in the arterial endothelium triggers the appearance of oxLDL [87]. In addition, this process drives an ROS-induced ROS mechanism, increasing mitochondrial ROS and promoting atherosclerosis progression [88]. An interesting study has shown that the accumulation of ROS also promotes DNA fragmentation and increases monocytes’ apoptosis in normocholesterolemic old mice, which is worsened in age-matched atherosclerotic mice, indicating that increased ROS may promote the aggravation of age-related atherosclerosis [89]. In addition, the aggregation of LDL-C in the arterial wall induces ROS production and enhances atherosclerosis progression in hypercholesterolemic mice [90]. Moreover, mice with transplanted bone marrow with mitochondrial dysfunction showed increased ROS levels and apoptotic cells, which contributed to the development of pro-atherosclerotic aortic lesions [91][92].
There are several factors related with ROS production in atherosclerosis, including sex, age, exercise, diet, obesity, smoking, hypertension, diabetes, and hyperlipidemia. Although ROS could act as the second messenger in various cellular pathways, their accumulation can cause the activation of inflammatory cytokines [93]. Moreover, severe DNA damage caused by excessive ROS overactivates the poly(Adenosine diphosphate (ADP)-ribose) polymerase 1 (PARP1) pathway, which leads to the functional impairment or death of VSMCs and vascular endothelial cells (VECs) through ATP and nicotinamide adenine dinucleotide (NAD+) depletion and, consequently, increases inflammation. In addition, mitochondrial malfunction results in ROS overproduction, which induces the oxidation of lipids, nucleic acids, and proteins, which eventually leads to severe cellular damage. Enhanced ROS production causes endothelial dysfunction, vascular inflammation, and accumulation of oxLDL in the arterial wall, which are responsible for the formation of the early plaque and its growth [94]. Taken together, these findings indicate that mitochondrial dysfunction, in combination with oxLDL, originates a continuous cycle associated with inflammation that will eventually lead to atheroma formation.
Apart from ROS, mitochondria are also able to release mitochondrial-derived damage-associated molecular patterns (mito-DAMPs), which show immunogenic capacity when misplaced or imbalanced. Mito-DAMPs are known as early inflammatory modulators in response to cellular stress, promoting the chemoattraction of immune cells [95]. Although mito-DAMPs could have a positive effect on tissue injury recovery [96], aberrant and chronical mito-DAMP release could lead to severe mitochondrial dysfunction and continuous inflammatory processes, leading to pathological disorders [95][96][97].
One proposed mitochondrial mechanism to enhance cell survival and reduce atherosclerotic damage is the release of humanin, a prominent member of a newly discovered family of mitochondrial-derived peptides expressed from an open reading frame of mitochondrial 16S rRNA. This mitochondrial-encoded peptide has been shown to play a role in preventing cell death among various tissues [98][99][100], including the endothelium [101]. Zacharias et al. demonstrated that humanin is expressed in the endothelium, smooth muscles, and macrophages during atherosclerosis and that its administration ex vivo results in decreased ROS production and apoptosis after oxLDL exposure of human aortic endothelial cells [102]. They also showed that humanin exerts a protective effect on endothelial function and atherosclerotic progression in ApoE-deficient mice [103]. The specific signal leading to a higher expression of humanin in atherosclerosis is unknown. Since apoptosis is a natural process in late-stage atherosclerosis contributing to the formation of a necrotic core and unstable plaque, the expression of humanin might be a defense mechanism to slow down the progression of the disease. Although unstable plaques present higher humanin levels, this compensatory response may not be sufficient to withstand sustained damage and, consequently, eventual ischemic events might arise [102].
3.1. Focusing on the Endothelial Origin of Atherosclerosis
Endothelial cells keep vascular homeostasis mostly by regulating vasodilation, platelet activation, leukocyte adhesion, and VSMC proliferation and migration [104]. Endothelial alteration is the first phase in early atherosclerosis (Figure 2), characterized by reduced NO secretion and synthesis [105]. NO prevents the expression of endothelium adhesion molecules and chemokines, as well as inhibits platelets’ activation and aggregation. These phenomena suggest that endothelial NO plays an important anti-inflammatory and anti-thrombotic role [106].
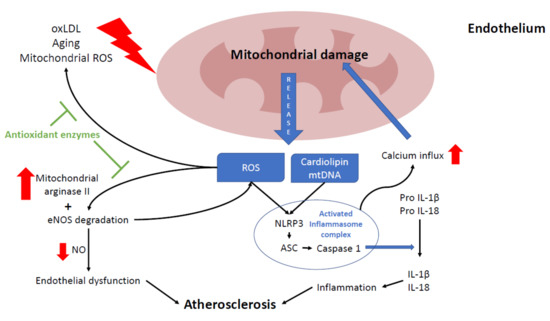
Figure 2. Relationship between mitochondria and atherosclerosis in the endothelium. Mitochondria play a key role in endothelium function, participating in several processes, such as nitric acid production, intracellular signaling, and cell death. Excessive reactive oxygen species (ROS) production leads to endothelium senescence, apoptosis, and atherosclerosis progression. Mitochondria can be damaged by oxLDL, ROS (mostly from mitochondrial origin), and the aging process. Damaged mitochondria release several mitochondrial components such as mtROS, cardiolipin and mtDNA which induce nod like receptor family pyrin domain containing 3 (NLRP3) inflammasome activation, leading to inflammation by increasing interleukin 1β and 18 maturation. Chronic NLRP3 activation will eventually lead to more mitochondrial damage by promoting mitochondrial calcium influx. Also, higher ROS levels disrupt the NO balance by boosting mitochondrial arginase II activity and causing eNOS degradation. In endothelium, the reduction of NO levels may also promote endothelial dysfunction and atherosclerosis. Most of these ROS-derived alterations are prevented by antioxidants enzymes (green block arrows).
In the endothelium, NO is mostly synthesized by the conversion of L-arginine into L-citrulline by endothelial nitric oxide synthase (eNOS). NO levels are mainly regulated by intracellular arginine levels, which in turn are controlled by mitochondrial arginase II. Therefore, NO requires a healthy MRC function to maintain appropriate levels [107]. Reduced NO levels in the cell are mainly caused by eNOS degradation due to high levels of ROS and loss of mitochondrial membrane potential (ΔΨm) [105][106]. In addition, eNOS inhibition contributes to more ROS production, given that it is also necessary for mitochondrial biogenesis [108], thus damaging endothelial cells and promoting the development of atherosclerosis [109]. In mice, the overexpression of mitochondrial arginase II in endothelial cells reduces NO levels [110]. The loss of this equilibrium was also reported in the human endothelium with high levels of oxLDL, which cause an increase in arginase II activity [111].
The peroxisome proliferator-activated receptor γ (PPARγ) helper activator-1α (PPARγ coactivator-1α (PGC-1α)) is one of the most important regulators of mitochondrial biosynthesis in most cells, including endothelial cells [112], and it is considered a key protein in mitochondrial homeostasis. In addition, PGC-1α controls the activity of mitochondrial transcript factors A and B, which coordinate mtDNA expression [113]. PGC-1α expression inhibits the damage of endothelium-dependent vasodilation induced by fatty acids and restores the function of NO. Won et al. [114] revealed that PGC-1α restored endothelial cell fatty acid oxidation, optimizing the ATP/ADP translocator activity and increasing ATP synthesis. These effects allow the control of ROS levels, maintaining endothelial cells’ activity and stress resistance. In fact, the overexpression of PGC-1α promotes NO production and both independently inhibit ROS generation, thereby delaying atherosclerosis appearance. The most upstream pro-inflammatory effector in atherosclerosis is NF-kB [115], and the presence of PGC-1α can decrease the activity of NF-kB and tumor necrosis factor α (TNFα), consequently blocking oxLDL-related inflammation, regulating vascular endothelial growth factor-1 (VEGF1) expression, and stimulating angiogenesis [105]. In conclusion, the activity of PGC-1α prevents apoptosis, restricts inflammatory activation, and increases NO synthesis [113].
Recently, the mitochondrial production of peroxynitrite (ONOO−), a nitrogen reactive species (RNS), has been gaining relevance in cancer, innate immunity, vascular diseases, and inflammation [116]. In the endothelium, the presence of this RNS is regulated by thioredoxin reductase 2 (TrxR2) via the steady-state concentration of ONOO−, the reaction product of superoxide radical and nitric oxide, and the integrity of the vascular system [117]. Kameritsch et al. showed that mice with endothelial deletion of the Trxrd2 gene develop increased vascular stiffness, hypertrophy of the vascular wall, and renal dysfunction [118]. In addition, increased ONOO− production was detected in LPS-treated mice and this overproduction of ONOO− was proportional to the developing progression of inflammation [119]. Impaired NO production was also related with enhanced ONOO− production [120]. All of these factors taken together, a high concentration of ONOO− has been directly linked with severe atherosclerosis damage [121].
3.2. Mitochondria and NLRP3 Inflammasome
Early evidence supporting the relationship between mitochondria and the NLRP3 inflammasome was reported by Zhou et al. [17], demonstrating that mitochondrial ROS are NLRP3 activators. Therefore, antioxidant compounds could block NLRP3 inflammasome assembly and ameliorate inflammation [122]. In addition, impaired mitophagy, a cellular process implicated in mitochondrial renewal, enhances mitochondrial damage and the release of ROS, mtDNA, and K+ into the cytoplasm, which promotes NLRP3 inflammasome activation. In fact, NRLP3 protein can interact directly with released mtDNA, initiating the inflammatory process [19]. Taken together, these findings support the hypothesis that impaired mitochondria could activate inflammation through NLRP3 in a direct way [123]. However, it is still unknown how NLRP3 activators can also induce mitochondrial damage. It is thought that they may alter intracellular Ca2+ homeostasis. One example is ATP, a canonical NLRP3 activator that can induce Ca2+ influx and promote the production of mitochondrial ROS and the loss of ΔΨm [124]. Furthermore, K+ efflux, caused by mitochondrial damage, can mediate the influx of Ca2+, resulting in a loss of mitochondrial Ca2+ homeostasis [125]. However, K+ efflux can also activate NLRP3 inflammasome independently of Ca2+ signaling [126]. New studies are required to evaluate the relevance of Ca2+ flux in NLRP3 inflammasome activation and mitochondrial dysfunction.
NLRP3 inflammasome can also be activated through the presence of bacterial molecules, such as N-acetylglucosamine, which are able to inhibit and dissociate the glycolytic enzyme hexokinase. Hexokinase is associated with the voltage-dependent anion channel (VDAC) at the mitochondrial outer membrane [127]. The VDAC regulates mitochondrial ROS production [128], can release large molecules (including mtDNA) into the cytosol [129], and is localized to cardiolipin-rich regions an NLRP3 activator [130]. The interaction of hexokinase with VDAC protects cells from mitochondrial ROS production [128] and inhibits the sustained opening of the mitochondrial permeability transition pore (MPTP) [129]. In addition, metabolic perturbations that inhibit hexokinase function, such as treatment with glucose-6-phosphate, 2-deoxyglucose, or citrate, all lead to inflammasome activation [131]. This evidence indicates the close relationship between metabolic alterations affecting hexokinase function and localization and inflammatory processes.
In contrast, several studies have reported another link between mitochondrial damage and NLRP3 activation. Thus, Yu et al. suggested that mitochondrial damage could be a side effect of inflammasome activation and, thereby, a consequence rather than a cause [132]. Additionally, the specific mitochondrial ROS activation mechanism in NLRP3 is still unknown. Despite the fact that ROS scavengers such as N-acetyl-cysteine (NAC) reduce the transcription of NLRP3 and pro-IL-1β, they have no apparent effect on mitochondrial ROS [133]. Furthermore, ROS generation was not affected in NLRP3−/− cells in response to LPS and ATP treatment, although NLRP3 deficiency was able to prevent mitochondrial depolarization [19]. Nevertheless, we are still far from establishing a clear connection between mitochondria and NLRP3.
Mitochondria themselves can also contribute to NLRP3 inflammasome formation by acting as an assembly platform. Specifically, MAVS and mitofusin 2 (Mfn2) have been proposed to recruit the NLRP3 protein to mitochondria in response to viral infection or non-mitochondrial NLRP3 activators. In its native form, most of the NLRP3 protein resides on the endoplasmic reticulum (ER). Upon stimulation with NLRP3 inducers, NLRP3 and ASC colocalize with mitochondria-associated ER membranes (MAMs) in the perinuclear space [134]. MAMs are essential in the initiation and regulation of the innate immune system, which includes inflammation [135], in addition to Ca2+ signaling [136]. Zhang et al. [134] described the following process: (1) After NLRP3 inflammasome activation, mitochondria cluster around the Golgi apparatus. (2) Diacylglycerol (DAG) starts to accumulate in Golgi and recruits protein kinase D (PKD). (3) PKD contributes to ASC oligomerization and NLRP3 phosphorylation and release from MAMs. (4) A new, fully mature NLRP3 inflammasome is released into the cytoplasm. To prove this process, PKD inactivation causes the retention of NLRP3 protein at MAMs adjacent to Golgi apparatus and reduces NLRP3 inflammasome assembly and activation. On the other hand, overexpression of PKD leads to NLRP3 inflammasome overactivation and IL-1β release without stimulation [134].
Mitochondria are a continuous and dynamic network that maintains an equilibrium between fusion, fission, repair, sequestration, degradation via mitophagy or mitophagy-independent mechanisms, and biogenesis [137][138]. Extracellular ATP, a well-known NLRP3 activator, binds to the P2X7 receptor, inducing K+ efflux, which could cause mitochondrial disruption [125]. In turn, mitochondrial damage could lead to the release of molecules (including mitochondrial ROS and mtDNA) that trigger NLRP3 inflammasome assembly [139]. As mitophagy allows the elimination of damaged mitochondria, a healthy rate of mitochondrial renewal will inhibit NLRP3 activation [18]. In contrast, mitophagy impairment by the depletion of autophagic proteins, such as microtubule-associated proteins 1A/1B light chain 3B II (LC3B-II) and Beclin-1, blocks the removal of malfunctioning mitochondria. Accumulation of damaged mitochondria promotes ROS generation and activation of the MPTP, which eventually leads to mtDNA release [19] and, as a consequence, NLRP3 inflammasome activation [20]. Furthermore, caspase-1 expression is highly decreased in LPS-primed murine phagocytes treated with ethidium bromide, which diminishes mtDNA copy number [19]. Likewise, NLRP3 inflammasome activation promotes cytosolic mtDNA release, which is abolished in LPS-primed NLRP3−/− macrophages in response to pro-inflammatory signals [140].
Many NLRP3 activators, such as oxLDL, induce alterations in ΔΨm, which in turn leads to an overproduction of mitochondrial ROS [17] and activation of Ca2+ signaling [141]. Furthermore, the inhibition of Complex I by rotenone induces the loss of ΔΨm, increases ROS production, and enhances NLRP3-dependent IL-1β secretion [17]. In atherosclerosis, NLRP3 activation has been correlated with mitochondrial impairment by in vitro experiments showing the involvement of lectin-type oxidized LDL receptor 1 (LOX-1), a major receptor for oxLDL, in promoting mitochondrial damage [142]. However, there is no evidence of this phenomenon in vivo. A study has reported that fatty-acid-mediated mitochondrial uncoupling facilitates NLRP3-independent interleukin 1α (IL-1α) release in vitro, but not that of IL-1β, and atherosclerosis development in vivo [143]. These findings are in disagreement with other reports regarding the role of IL-1β release upon inflammasome activation in atherosclerosis [54].
All the proposed mechanisms for NLRP3 inflammasome activation, such as oxidative stress, mitochondrial dysfunction [144], ER stress [145], and lysosome rupture [146], have been observed in atherosclerosis. In addition, exogenous Ca2+ has been proposed as a new inflammasome activator due to its high concentration in atherosclerotic plaques, specifically around necrotic regions [147]. Finally, mitochondrial dysfunction has been demonstrated in dyslipidemia-related genetic disorders such as familial hypercholesterolemia [148][149].
3.3. Mitochondrial Mutations and Atherogenesis
The appearance of atherosclerosis in mitochondrial diseases may result from a primary pathological mechanism or as a consequence of a secondary mechanism associated with diabetes, arterial hypertension, or hyperlipidemia [150]. Most frequently, patients harboring mutations in mtDNA may develop primary mitochondrial atherosclerosis. Although the precise mechanism is not clear, it is believed that primary mitochondrial atherosclerosis results from increased oxidative stress, mitophagy alterations, energy deficiency, accumulation of toxic metabolites, or NLRP3 inflammasome activation. Thus, atherosclerosis in mitochondrial disorders may occur even in the absence of recognized atherosclerosis risk factors, suggesting that atherosclerosis can be a primary consequence of mitochondrial defect. However, there is still no direct evidence relating mitochondrial dysfunction to atherosclerosis progression [150].
Mitochondria could be directly linked to atherosclerosis in several ways. For this reason, mitochondrial dysfunction or excessive ROS caused by mitochondrial genetic mutations could promote atherogenesis [150]. In fact, it has been demonstrated that some mitochondrial mutations may lead to chronic inflammatory processes associated with NLRP3 activation [151], a key pathogenic factor in atherosclerosis development. This group of genetic alterations and the associated mitochondrial dysfunction may enhance the mitochondrial release of mtDNA and other mitochondrial DAMPs, initiating an inflammatory response through NLRP3 inflammasome activation [152].
Mitochondrial ROS have been suggested to be the origin of an increased mutation rate in mtDNA, one that is up to 15 times higher than that of nuclear DNA [153]. On the other hand, the presence of multiple copies of mtDNA in single cells explains the phenomenon of heteroplasmy, which refers to the variable proportion of wild-type and mutant mtDNA copies within the cell or the tissue [154]. This proportion between different mtDNA varies with aging and is dependent on the cell type and the tissue [155]. The assessment of the mtDNA heteroplasmic mutation load, as well as the mtDNA copy number, is a plausible way to explain the focality of atherosclerotic lesions [156][157]. Thus, cells possessing the level of heteroplasmy exceeding a certain threshold may exhibit mitochondrial dysfunction, whereas in the adjacent cells with a lower mutational load, the respiratory chain remains fully functional [158]. A similar mosaic pattern can be observed in atherosclerosis, where healthy areas of arterial wall coexist with regions bearing different types of atherosclerotic lesions [159][160].
Nowadays, several studies assume a connection between mtDNA mutations and atherosclerosis [161][162][163]. These studies were performed using leukocytes and arterial wall samples from atherosclerotic human patients. Most mutations were related to mitochondrial ribosomes, mitochondrial transfer RNA, and various mitochondrial-encoded respiratory complex subunits. It has been proposed that the presence of these mutations promotes mitochondrial dysfunction and therefore ROS production, which enhances the appearance of atherosclerotic plaques and increases the thickness of the intima and medial layers in carotid arteries [162]. Interestingly, Sazonova et al. discovered the presence of several mtDNA mutations associated with patients presenting with carotid atherosclerosis as well as two single-nucleotide substitutions that negatively correlate with atherosclerotic lesions [162]. It has been proposed that these mutations can be biomarkers for assessing predisposition to this disease.
Despite the available evidence, there is no established consensus on the importance of heteroplasmy in atherosclerosis [164]. The main hypothesis suggests that a primary defect of the respiratory chain or OXPHOS system may be associated with reduced energy production that can ultimately lead to the collapse of cell energy metabolism. Uncoupling of the electron transfer from ATP synthesis results in the excess generation of ROS, leading to widespread cellular injury and vascular damage [165]. In addition, ROS overproduction may enhance the mtDNA mutation rate and, as a consequence, further increase ROS production [166].
One of the mechanisms that enable cells to cope with heteroplasmy and mitochondrial dysfunction is mitophagy. Mitophagy surveils the mitochondrial population, removing unnecessary and/or impaired organelles. [167]. This process requires high energy and regulates NLRP3 inflammasome activation [18][168]. Therefore, the defective removal of damaged mitochondria leads to the hyperactivation of inflammatory signaling pathways and, subsequently, to chronic local or systemic inflammation [169][170], which may result in focal or generalized atherosclerosis [171]. In agreement with this assumption, in vitro inhibition of mitophagy in a primary culture of human monocyte-derived macrophages increased pro-inflammatory response in the form of up-regulation of the IL-1β gene [172].
However, it is not clear whether it is the increased mitochondrial ROS caused by atherosclerosis throughout lifetime that increases somatic mtDNA mutations or if it is previous maternally inherited mtDNA mutations that promote atherosclerosis [164]. Overall, the mechanism behind the role of heteroplasmic mtDNA variants in the development of atherosclerosis is still not completely understood.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines9030258
References
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560.
- Chien, Y.; Rosal, K.; Chung, B.C. Function of CYP11A1 in the mitochondria. Mol. Cell Endocrinol. 2017, 441, 55–61.
- Bravo-Sagua, R.; Parra, V.; Lopez-Crisosto, C.; Diaz, P.; Quest, A.F.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. Compr Physiol 2017, 7, 623–634.
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.V. Mitochondria and Iron: Current questions. Expert Rev. Hematol. 2017, 10, 65–79.
- Liu, P.S.; Ho, P.C. Mitochondria: A master regulator in macrophage and T cell immunity. Mitochondrion 2018, 41, 45–50.
- Blajszczak, C.; Bonini, M.G. Mitochondria targeting by environmental stressors: Implications for redox cellular signaling. Toxicology 2017, 391, 84–89.
- Jeong, S.Y.; Seol, D.W. The role of mitochondria in apoptosis. BMB Rep. 2008, 41, 11–22.
- Kolmychkova, K.I.; Zhelankin, A.V.; Karagodin, V.P.; Orekhov, A.N. Mitochondria and inflammation. Patol. Fiziol. Eksp. Ter. 2016, 60, 114–121.
- Kuprash, D.V.; Nedospasov, S.A. Molecular and Cellular Mechanisms of Inflammation. Biochemistry (Mosc) 2016, 81, 1237–1239.
- Escamilla-Tilch, M.; Filio-Rodriguez, G.; Garcia-Rocha, R.; Mancilla-Herrera, I.; Mitchison, N.A.; Ruiz-Pacheco, J.A.; Sanchez-Garcia, F.J.; Sandoval-Borrego, D.; Vazquez-Sanchez, E.A. The interplay between pathogen-associated and danger-associated molecular patterns: An inflammatory code in cancer? Immunol. Cell Biol. 2013, 91, 601–610.
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435.
- Nace, G.; Evankovich, J.; Eid, R.; Tsung, A. Dendritic cells and damage-associated molecular patterns: Endogenous danger signals linking innate and adaptive immunity. J. Innate Immun. 2012, 4, 6–15.
- Elieh Ali Komi, D.; Sharma, L.; Dela Cruz, C.S. Chitin and Its Effects on Inflammatory and Immune Responses. Clin. Rev. Allergy Immunol 2018, 54, 213–223.
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820.
- Archibald, J.M. Endosymbiosis and Eukaryotic Cell Evolution. Curr. Biol. 2015, 25, R911–R921.
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107.
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225.
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-kappaB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910.
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230.
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414.
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557.
- Liu, B.; Gao, C. Regulation of MAVS activation through post-translational modifications. Curr. Opin. Immunol. 2018, 50, 75–81.
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498.
- Subramanian, N.; Natarajan, K.; Clatworthy, M.R.; Wang, Z.; Germain, R.N. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 2013, 153, 348–361.
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480.
- Brown, J.; Wang, H.; Hajishengallis, G.N.; Martin, M. TLR-signaling networks: An integration of adaptor molecules, kinases, and cross-talk. J. Dent. Res. 2011, 90, 417–427.
- Banoth, B.; Cassel, S.L. Mitochondria in innate immune signaling. Transl. Res. 2018, 202, 52–68.
- Mallat, Z.; Corbaz, A.; Scoazec, A.; Besnard, S.; Leseche, G.; Chvatchko, Y.; Tedgui, A. Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation 2001, 104, 1598–1603.
- Thomas, H.; Diamond, J.; Vieco, A.; Chaudhuri, S.; Shinnar, E.; Cromer, S.; Perel, P.; Mensah, G.A.; Narula, J.; Johnson, C.O.; et al. Global Atlas of Cardiovascular Disease 2000-2016: The Path to Prevention and Control. Glob. Heart 2018, 13, 143–163.
- Tietge, U.J. Hyperlipidemia and cardiovascular disease: Inflammation, dyslipidemia, and atherosclerosis. Curr Opin Lipidol 2014, 25, 94–95.
- Fisher, E.A.; Feig, J.E.; Hewing, B.; Hazen, S.L.; Smith, J.D. High-density lipoprotein function, dysfunction, and reverse cholesterol transport. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2813–2820.
- Boren, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020.
- Mestas, J.; Ley, K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc. Med. 2008, 18, 228–232.
- Li, Y.; C, G.Z.; Wang, X.H.; Liu, D.H. Progression of atherosclerosis in ApoE-knockout mice fed on a high-fat diet. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 3863–3867.
- Emini Veseli, B.; Perrotta, P.; De Meyer, G.R.A.; Roth, L.; Van der Donckt, C.; Martinet, W.; De Meyer, G.R.Y. Animal models of atherosclerosis. Eur. J. Pharmacol 2017, 816, 3–13.
- Libby, P.; Ridker, P.M.; Hansson, G.K.; Leducq Transatlantic Network on, A. Inflammation in atherosclerosis: From pathophysiology to practice. J. Am. Coll. Cardiol. 2009, 54, 2129–2138.
- Ridker, P.M. Residual inflammatory risk: Addressing the obverse side of the atherosclerosis prevention coin. Eur Heart J. 2016, 37, 1720–1722.
- Zhu, Y.; Xian, X.; Wang, Z.; Bi, Y.; Chen, Q.; Han, X.; Tang, D.; Chen, R. Research Progress on the Relationship between Atherosclerosis and Inflammation. Biomolecules 2018, 8, 80.
- Ahotupa, M. Oxidized lipoprotein lipids and atherosclerosis. Free Radic. Res. 2017, 51, 439–447.
- Johnson, J.L. Metalloproteinases in atherosclerosis. Eur. J. Pharmacol. 2017, 816, 93–106.
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866.
- Badimon, L.; Padro, T.; Vilahur, G. Atherosclerosis, platelets and thrombosis in acute ischaemic heart disease. Eur. Heart J. Acute Cardiovasc. Care 2012, 1, 60–74.
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426.
- Lu, A.; Wu, H. Structural mechanisms of inflammasome assembly. FEBS J. 2015, 282, 435–444.
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326 (Pt. 1), 1–16.
- Gu, Y.; Kuida, K.; Tsutsui, H.; Ku, G.; Hsiao, K.; Fleming, M.A.; Hayashi, N.; Higashino, K.; Okamura, H.; Nakanishi, K.; et al. Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science 1997, 275, 206–209.
- Rathinam, V.A.; Fitzgerald, K.A. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 2016, 165, 792–800.
- Haneklaus, M.; O’Neill, L.A. NLRP3 at the interface of metabolism and inflammation. Immunol. Rev. 2015, 265, 53–62.
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021.
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687.
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129.
- Frank, M.G.; Weber, M.D.; Watkins, L.R.; Maier, S.F. Stress-induced neuroinflammatory priming: A liability factor in the etiology of psychiatric disorders. Neurobiol. Stress 2016, 4, 62–70.
- Alexander, M.R.; Moehle, C.W.; Johnson, J.L.; Yang, Z.; Lee, J.K.; Jackson, C.L.; Owens, G.K. Genetic inactivation of IL-1 signaling enhances atherosclerotic plaque instability and reduces outward vessel remodeling in advanced atherosclerosis in mice. J. Clin. Invest. 2012, 122, 70–79.
- Libby, P. Interleukin-1 Beta as a Target for Atherosclerosis Therapy: Biological Basis of CANTOS and Beyond. J. Am. Coll. Cardiol. 2017, 70, 2278–2289.
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361.
- Klinkner, A.M.; Waites, C.R.; Kerns, W.D.; Bugelski, P.J. Evidence of foam cell and cholesterol crystal formation in macrophages incubated with oxidized LDL by fluorescence and electron microscopy. J. Histochem. Cytochem. 1995, 43, 1071–1078.
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820.
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131.
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128.
- Westerterp, M.; Fotakis, P.; Ouimet, M.; Bochem, A.E.; Zhang, H.; Molusky, M.M.; Wang, W.; Abramowicz, S.; la Bastide-van Gemert, S.; Wang, N.; et al. Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis, and Atherogenesis. Circulation 2018, 138, 898–912.
- Usui, F.; Shirasuna, K.; Kimura, H.; Tatsumi, K.; Kawashima, A.; Karasawa, T.; Hida, S.; Sagara, J.; Taniguchi, S.; Takahashi, M. Critical role of caspase-1 in vascular inflammation and development of atherosclerosis in Western diet-fed apolipoprotein E-deficient mice. Biochem. Biophys. Res. Commun. 2012, 425, 162–168.
- Gage, J.; Hasu, M.; Thabet, M.; Whitman, S.C. Caspase-1 deficiency decreases atherosclerosis in apolipoprotein E-null mice. Can. J. Cardiol 2012, 28, 222–229.
- Zheng, F.; Xing, S.; Gong, Z.; Mu, W.; Xing, Q. Silence of NLRP3 suppresses atherosclerosis and stabilizes plaques in apolipoprotein E-deficient mice. Mediators Inflamm. 2014, 2014, 507208.
- Bai, B.; Yang, Y.; Wang, Q.; Li, M.; Tian, C.; Liu, Y.; Aung, L.H.H.; Li, P.F.; Yu, T.; Chu, X.M. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis 2020, 11, 776.
- Galea, J.; Armstrong, J.; Gadsdon, P.; Holden, H.; Francis, S.E.; Holt, C.M. Interleukin-1 beta in coronary arteries of patients with ischemic heart disease. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1000–1006.
- Bevilacqua, M.P.; Pober, J.S.; Majeau, G.R.; Cotran, R.S.; Gimbrone, M.A., Jr. Interleukin 1 (IL-1) induces biosynthesis and cell surface expression of procoagulant activity in human vascular endothelial cells. J. Exp. Med. 1984, 160, 618–623.
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702.
- Baylis, R.A.; Gomez, D.; Mallat, Z.; Pasterkamp, G.; Owens, G.K. The CANTOS Trial: One Important Step for Clinical Cardiology but a Giant Leap for Vascular Biology. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e174–e177.
- Pavillard, L.E.; Marin-Aguilar, F.; Bullon, P.; Cordero, M.D. Cardiovascular diseases, NLRP3 inflammasome, and western dietary patterns. Pharmacol. Res. 2018, 131, 44–50.
- Zheng, F.; Xing, S.; Gong, Z.; Xing, Q. NLRP3 inflammasomes show high expression in aorta of patients with atherosclerosis. Heart Lung Circ. 2013, 22, 746–750.
- Shi, X.; Xie, W.L.; Kong, W.W.; Chen, D.; Qu, P. Expression of the NLRP3 Inflammasome in Carotid Atherosclerosis. J. Stroke Cerebrovasc. Dis. 2015, 24, 2455–2466.
- Paramel Varghese, G.; Folkersen, L.; Strawbridge, R.J.; Halvorsen, B.; Yndestad, A.; Ranheim, T.; Krohg-Sorensen, K.; Skjelland, M.; Espevik, T.; Aukrust, P.; et al. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. J. Am. Heart Assoc. 2016, 5.
- Afrasyab, A.; Qu, P.; Zhao, Y.; Peng, K.; Wang, H.; Lou, D.; Niu, N.; Yuan, D. Correlation of NLRP3 with severity and prognosis of coronary atherosclerosis in acute coronary syndrome patients. Heart Vessels 2016, 31, 1218–1229.
- Siasos, G.; Tsigkou, V.; Kosmopoulos, M.; Theodosiadis, D.; Simantiris, S.; Tagkou, N.M.; Tsimpiktsioglou, A.; Stampouloglou, P.K.; Oikonomou, E.; Mourouzis, K.; et al. Mitochondria and cardiovascular diseases-from pathophysiology to treatment. Ann. Transl. Med. 2018, 6, 256.
- Graves, S.M.; Xie, Z.; Stout, K.A.; Zampese, E.; Burbulla, L.F.; Shih, J.C.; Kondapalli, J.; Patriarchi, T.; Tian, L.; Brichta, L.; et al. Dopamine metabolism by a monoamine oxidase mitochondrial shuttle activates the electron transport chain. Nat. Neurosci. 2020, 23, 15–20.
- Kundu, T.K.; Velayutham, M.; Zweier, J.L. Aldehyde oxidase functions as a superoxide generating NADH oxidase: An important redox regulated pathway of cellular oxygen radical formation. Biochemistry 2012, 51, 2930–2939.
- Li, R.; Jia, Z.; Trush, M.A. Defining ROS in Biology and Medicine. React. Oxyg. Species (Apex) 2016, 1, 9–21.
- Schroder, K. NADPH oxidase-derived reactive oxygen species: Dosis facit venenum. Exp. Physiol. 2019, 104, 447–452.
- El-Benna, J.; Hurtado-Nedelec, M.; Marzaioli, V.; Marie, J.C.; Gougerot-Pocidalo, M.A.; Dang, P.M. Priming of the neutrophil respiratory burst: Role in host defense and inflammation. Immunol. Rev. 2016, 273, 180–193.
- Brandes, R.P.; Weissmann, N.; Schroder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226.
- Helmcke, I.; Heumuller, S.; Tikkanen, R.; Schroder, K.; Brandes, R.P. Identification of structural elements in Nox1 and Nox4 controlling localization and activity. Antioxid. Redox Signal 2009, 11, 1279–1287.
- Jha, J.C.; Watson, A.M.D.; Mathew, G.; de Vos, L.C.; Jandeleit-Dahm, K. The emerging role of NADPH oxidase NOX5 in vascular disease. Clin. Sci. 2017, 131, 981–990.
- Wang, Y.; Wang, G.Z.; Rabinovitch, P.S.; Tabas, I. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor-kappaB-mediated inflammation in macrophages. Circ. Res. 2014, 114, 421–433.
- Yeh, H.L.; Kuo, L.T.; Sung, F.C.; Yeh, C.C. Association between Polymorphisms of Antioxidant Gene (MnSOD, CAT, and GPx1) and Risk of Coronary Artery Disease. Biomed. Res. Int. 2018, 2018, 5086869.
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905.
- Velayutham, M.; Hemann, C.; Zweier, J.L. Removal of H(2)O(2) and generation of superoxide radical: Role of cytochrome c and NADH. Free Radic. Biol. Med. 2011, 51, 160–170.
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420.
- Yu, E.P.K.; Reinhold, J.; Yu, H.; Starks, L.; Uryga, A.K.; Foote, K.; Finigan, A.; Figg, N.; Pung, Y.F.; Logan, A.; et al. Mitochondrial Respiration Is Reduced in Atherosclerosis, Promoting Necrotic Core Formation and Reducing Relative Fibrous Cap Thickness. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2322–2332.
- Jacinto, T.A.; Meireles, G.S.; Dias, A.T.; Aires, R.; Porto, M.L.; Gava, A.L.; Vasquez, E.C.; Pereira, T.M.C.; Campagnaro, B.P.; Meyrelles, S.S. Increased ROS production and DNA damage in monocytes are biomarkers of aging and atherosclerosis. Biol. Res. 2018, 51, 33.
- Vilne, B.; Skogsberg, J.; Foroughi Asl, H.; Talukdar, H.A.; Kessler, T.; Bjorkegren, J.L.M.; Schunkert, H. Network analysis reveals a causal role of mitochondrial gene activity in atherosclerotic lesion formation. Atherosclerosis 2017, 267, 39–48.
- Blanc, J.; Alves-Guerra, M.C.; Esposito, B.; Rousset, S.; Gourdy, P.; Ricquier, D.; Tedgui, A.; Miroux, B.; Mallat, Z. Protective role of uncoupling protein 2 in atherosclerosis. Circulation 2003, 107, 388–390.
- Arsenijevic, D.; Onuma, H.; Pecqueur, C.; Raimbault, S.; Manning, B.S.; Miroux, B.; Couplan, E.; Alves-Guerra, M.C.; Goubern, M.; Surwit, R.; et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet. 2000, 26, 435–439.
- El-Kenawi, A.; Ruffell, B. Inflammation, ROS, and Mutagenesis. Cancer Cell 2017, 32, 727–729.
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127.
- An, P.; Wei, L.L.; Zhao, S.; Sverdlov, D.Y.; Vaid, K.A.; Miyamoto, M.; Kuramitsu, K.; Lai, M.; Popov, Y.V. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat. Commun. 2020, 11, 2362.
- Qualls, A.E.; Southern, W.M.; Call, J.A. Mitochondria-Cytokine Crosstalk Following Skeletal Muscle Injury and Disuse: A Mini-Review. Am. J. Physiol. Cell Physiol. 2021.
- Chakraborty, K.; Raundhal, M.; Chen, B.B.; Morse, C.; Tyurina, Y.Y.; Khare, A.; Oriss, T.B.; Huff, R.; Lee, J.S.; St Croix, C.M.; et al. The mito-DAMP cardiolipin blocks IL-10 production causing persistent inflammation during bacterial pneumonia. Nat. Commun. 2017, 8, 13944.
- Colon, E.; Strand, M.L.; Carlsson-Skwirut, C.; Wahlgren, A.; Svechnikov, K.V.; Cohen, P.; Soder, O. Anti-apoptotic factor humanin is expressed in the testis and prevents cell-death in leydig cells during the first wave of spermatogenesis. J. Cell Physiol. 2006, 208, 373–385.
- Niikura, T.; Tajima, H.; Kita, Y. Neuronal cell death in Alzheimer’s disease and a neuroprotective factor, humanin. Curr Neuropharmacol. 2006, 4, 139–147.
- Muzumdar, R.H.; Huffman, D.M.; Calvert, J.W.; Jha, S.; Weinberg, Y.; Cui, L.; Nemkal, A.; Atzmon, G.; Klein, L.; Gundewar, S.; et al. Acute humanin therapy attenuates myocardial ischemia and reperfusion injury in mice. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1940–1948.
- Widmer, R.J.; Flammer, A.J.; Herrmann, J.; Rodriguez-Porcel, M.; Wan, J.; Cohen, P.; Lerman, L.O.; Lerman, A. Circulating humanin levels are associated with preserved coronary endothelial function. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H393–H397.
- Zacharias, D.G.; Kim, S.G.; Massat, A.E.; Bachar, A.R.; Oh, Y.K.; Herrmann, J.; Rodriguez-Porcel, M.; Cohen, P.; Lerman, L.O.; Lerman, A. Humanin, a cytoprotective peptide, is expressed in carotid atherosclerotic [corrected] plaques in humans. PLoS ONE 2012, 7, e31065.
- Oh, Y.K.; Bachar, A.R.; Zacharias, D.G.; Kim, S.G.; Wan, J.; Cobb, L.J.; Lerman, L.O.; Cohen, P.; Lerman, A. Humanin preserves endothelial function and prevents atherosclerotic plaque progression in hypercholesterolemic ApoE deficient mice. Atherosclerosis 2011, 219, 65–73.
- Szewczyk, A.; Jarmuszkiewicz, W.; Koziel, A.; Sobieraj, I.; Nobik, W.; Lukasiak, A.; Skup, A.; Bednarczyk, P.; Drabarek, B.; Dymkowska, D.; et al. Mitochondrial mechanisms of endothelial dysfunction. Pharmacol. Rep. 2015, 67, 704–710.
- Kadlec, A.O.; Chabowski, D.S.; Ait-Aissa, K.; Gutterman, D.D. Role of PGC-1alpha in Vascular Regulation: Implications for Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1467–1474.
- Pircher, A.; Treps, L.; Bodrug, N.; Carmeliet, P. Endothelial cell metabolism: A novel player in atherosclerosis? Basic principles and therapeutic opportunities. Atherosclerosis 2016, 253, 247–257.
- Mantzarlis, K.; Tsolaki, V.; Zakynthinos, E. Role of Oxidative Stress and Mitochondrial Dysfunction in Sepsis and Potential Therapies. Oxid. Med. Cell Longev. 2017, 2017, 5985209.
- Huang, P.L. eNOS, metabolic syndrome and cardiovascular disease. Trends Endocrinol. Metab. 2009, 20, 295–302.
- Hong, F.F.; Liang, X.Y.; Liu, W.; Lv, S.; He, S.J.; Kuang, H.B.; Yang, S.L. Roles of eNOS in atherosclerosis treatment. Inflamm. Res. 2019, 68, 429–441.
- Vaisman, B.L.; Andrews, K.L.; Khong, S.M.; Wood, K.C.; Moore, X.L.; Fu, Y.; Kepka-Lenhart, D.M.; Morris, S.M., Jr.; Remaley, A.T.; Chin-Dusting, J.P. Selective endothelial overexpression of arginase II induces endothelial dysfunction and hypertension and enhances atherosclerosis in mice. PLoS ONE 2012, 7, e39487.
- Wang, Q.; Zhao, T.; Zhang, W.; Yu, W.; Liu, B.; Wang, Z.; Qiao, W.; Lu, Q.; Wang, A.; Zhang, M. Poly (ADP-Ribose) Polymerase 1 Mediated Arginase II Activation Is Responsible for Oxidized LDL-Induced Endothelial Dysfunction. Front. Pharmacol. 2018, 9, 882.
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. 2012, 23, 459–466.
- Kluge, M.A.; Fetterman, J.L.; Vita, J.A. Mitochondria and endothelial function. Circ. Res. 2013, 112, 1171–1188.
- Won, J.C.; Park, J.Y.; Kim, Y.M.; Koh, E.H.; Seol, S.; Jeon, B.H.; Han, J.; Kim, J.R.; Park, T.S.; Choi, C.S.; et al. Peroxisome proliferator-activated receptor-gamma coactivator 1-alpha overexpression prevents endothelial apoptosis by increasing ATP/ADP translocase activity. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 290–297.
- Coto, E.; Reguero, J.R.; Avanzas, P.; Pascual, I.; Martin, M.; Hevia, S.; Moris, C.; Diaz-Molina, B.; Lambert, J.L.; Alonso, B.; et al. Gene variants in the NF-KB pathway (NFKB1, NFKBIA, NFKBIZ) and risk for early-onset coronary artery disease. Immunol. Lett. 2019, 208, 39–43.
- Szabo, C.; Ischiropoulos, H.; Radi, R. Peroxynitrite: Biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov. 2007, 6, 662–680.
- Yoshioka, J. Thioredoxin Reductase 2 (Txnrd2) Regulates Mitochondrial Integrity in the Progression of Age-Related Heart Failure. J. Am. Heart Assoc. 2015, 4.
- Kameritsch, P.; Singer, M.; Nuernbergk, C.; Rios, N.; Reyes, A.M.; Schmidt, K.; Kirsch, J.; Schneider, H.; Muller, S.; Pogoda, K.; et al. The mitochondrial thioredoxin reductase system (TrxR2) in vascular endothelium controls peroxynitrite levels and tissue integrity. Proc. Natl. Acad. Sci. USA 2021, 118.
- Wang, Z.; Wang, W.; Wang, P.; Song, X.; Mao, Z.; Liu, Z. Highly Sensitive Near-Infrared Imaging of Peroxynitrite Fluxes in Inflammation Progress. Anal. Chem. 2021, 93, 3035–3041.
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848.
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, indicators, risk factors and new hopes. Int. J. Prev. Med. 2014, 5, 927–946.
- Ip, W.K.; Medzhitov, R. Macrophages monitor tissue osmolarity and induce inflammatory response through NLRP3 and NLRC4 inflammasome activation. Nat. Commun. 2015, 6, 6931.
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immuno.l 2013, 14, 454–460.
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287.
- Yaron, J.R.; Gangaraju, S.; Rao, M.Y.; Kong, X.; Zhang, L.; Su, F.; Tian, Y.; Glenn, H.L.; Meldrum, D.R. K(+) regulates Ca(2+) to drive inflammasome signaling: Dynamic visualization of ion flux in live cells. Cell Death Dis. 2015, 6, e1954.
- Katsnelson, M.A.; Rucker, L.G.; Russo, H.M.; Dubyak, G.R. K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J. Immunol. 2015, 194, 3937–3952.
- Wolf, A.J.; Reyes, C.N.; Liang, W.; Becker, C.; Shimada, K.; Wheeler, M.L.; Cho, H.C.; Popescu, N.I.; Coggeshall, K.M.; Arditi, M.; et al. Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell 2016, 166, 624–636.
- da-Silva, W.S.; Gomez-Puyou, A.; de Gomez-Puyou, M.T.; Moreno-Sanchez, R.; De Felice, F.G.; de Meis, L.; Oliveira, M.F.; Galina, A. Mitochondrial bound hexokinase activity as a preventive antioxidant defense: Steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J. Biol. Chem. 2004, 279, 39846–39855.
- Rasola, A.; Sciacovelli, M.; Pantic, B.; Bernardi, P. Signal transduction to the permeability transition pore. FEBS Lett. 2010, 584, 1989–1996.
- Sun, Y.; Vashisht, A.A.; Tchieu, J.; Wohlschlegel, J.A.; Dreier, L. Voltage-dependent anion channels (VDACs) recruit Parkin to defective mitochondria to promote mitochondrial autophagy. J. Biol. Chem. 2012, 287, 40652–40660.
- Hughes, M.M.; O’Neill, L.A.J. Metabolic regulation of NLRP3. Immunol. Rev. 2018, 281, 88–98.
- Yu, J.; Nagasu, H.; Murakami, T.; Hoang, H.; Broderick, L.; Hoffman, H.M.; Horng, T. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 15514–15519.
- Bauernfeind, F.; Bartok, E.; Rieger, A.; Franchi, L.; Nunez, G.; Hornung, V. Cutting edge: Reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J. Immunol. 2011, 187, 613–617.
- Zhang, Z.; Meszaros, G.; He, W.T.; Xu, Y.; de Fatima Magliarelli, H.; Mailly, L.; Mihlan, M.; Liu, Y.; Puig Gamez, M.; Goginashvili, A.; et al. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J. Exp. Med. 2017, 214, 2671–2693.
- Misawa, T.; Takahama, M.; Saitoh, T. Mitochondria-Endoplasmic Reticulum Contact Sites Mediate Innate Immune Responses. Adv. Exp. Med. Biol 2017, 997, 187–197.
- de Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610.
- Suarez-Rivero, J.M.; Villanueva-Paz, M.; de la Cruz-Ojeda, P.; de la Mata, M.; Cotan, D.; Oropesa-Avila, M.; de Lavera, I.; Alvarez-Cordoba, M.; Luzon-Hidalgo, R.; Sanchez-Alcazar, J.A. Mitochondrial Dynamics in Mitochondrial Diseases. Diseases 2016, 5, 1.
- Sterky, F.H.; Lee, S.; Wibom, R.; Olson, L.; Larsson, N.G. Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 12937–12942.
- Elliott, E.I.; Sutterwala, F.S. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev. 2015, 265, 35–52.
- Park, S.; Won, J.H.; Hwang, I.; Hong, S.; Lee, H.K.; Yu, J.W. Defective mitochondrial fission augments NLRP3 inflammasome activation. Sci. Rep. 2015, 5, 15489.
- Triantafilou, K.; Hughes, T.R.; Triantafilou, M.; Morgan, B.P. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J. Cell Sci. 2013, 126, 2903–2913.
- Ding, Z.; Liu, S.; Wang, X.; Dai, Y.; Khaidakov, M.; Deng, X.; Fan, Y.; Xiang, D.; Mehta, J.L. LOX-1, mtDNA damage, and NLRP3 inflammasome activation in macrophages: Implications in atherogenesis. Cardiovasc. Res. 2014, 103, 619–628.
- Freigang, S.; Ampenberger, F.; Weiss, A.; Kanneganti, T.D.; Iwakura, Y.; Hersberger, M.; Kopf, M. Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent IL-1alpha and sterile vascular inflammation in atherosclerosis. Nat. Immunol. 2013, 14, 1045–1053.
- Victor, V.M.; Apostolova, N.; Herance, R.; Hernandez-Mijares, A.; Rocha, M. Oxidative stress and mitochondrial dysfunction in atherosclerosis: Mitochondria-targeted antioxidants as potential therapy. Curr. Med. Chem. 2009, 16, 4654–4667.
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N.; Bobryshev, Y.V. Role of endoplasmic reticulum stress in atherosclerosis and diabetic macrovascular complications. Biomed. Res. Int. 2014, 2014, 610140.
- Yuan, X.M.; Li, W.; Brunk, U.T.; Dalen, H.; Chang, Y.H.; Sevanian, A. Lysosomal destabilization during macrophage damage induced by cholesterol oxidation products. Free Radic. Biol. Med. 2000, 28, 208–218.
- de Zoete, M.R.; Palm, N.W.; Zhu, S.; Flavell, R.A. Inflammasomes. Cold Spring Harb. Perspect. Biol. 2014, 6, a016287.
- Suarez-Rivero, J.M.; de la Mata, M.; Pavon, A.D.; Villanueva-Paz, M.; Povea-Cabello, S.; Cotan, D.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Ybot-Gonzalez, P.; Salas, J.J.; et al. Intracellular cholesterol accumulation and coenzyme Q10 deficiency in Familial Hypercholesterolemia. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3697–3713.
- Suarez-Rivero, J.M.; Pastor-Maldonado, C.J.; de la Mata, M.; Villanueva-Paz, M.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Suarez-Carrillo, A.; Talaveron-Rey, M.; Munuera, M.; et al. Atherosclerosis and Coenzyme Q10. Int J. Mol. Sci. 2019, 20, 5195.
- Finsterer, J. Atherosclerosis Can Be Mitochondrial: A Review. Cureus 2020, 12, e6987.
- Cordero, M.D.; Alcocer-Gomez, E.; Marin-Aguilar, F.; Rybkina, T.; Cotan, D.; Perez-Pulido, A.; Alvarez-Suarez, J.M.; Battino, M.; Sanchez-Alcazar, J.A.; Carrion, A.M.; et al. Mutation in cytochrome b gene of mitochondrial DNA in a family with fibromyalgia is associated with NLRP3-inflammasome activation. J. Med. Genet. 2016, 53, 113–122.
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799.
- Yang, Y.; Karakhanova, S.; Hartwig, W.; D’Haese, J.G.; Philippov, P.P.; Werner, J.; Bazhin, A.V. Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy. J. Cell Physiol. 2016, 231, 2570–2581.
- Stefano, G.B.; Bjenning, C.; Wang, F.; Wang, N.; Kream, R.M. Mitochondrial Heteroplasmy. Adv. Exp. Med. Biol 2017, 982, 577–594.
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542.
- Liu, L.P.; Cheng, K.; Ning, M.A.; Li, H.H.; Wang, H.C.; Li, F.; Chen, S.Y.; Qu, F.L.; Guo, W.Y. Association between peripheral blood cells mitochondrial DNA content and severity of coronary heart disease. Atherosclerosis 2017, 261, 105–110.
- Ashar, F.N.; Zhang, Y.; Longchamps, R.J.; Lane, J.; Moes, A.; Grove, M.L.; Mychaleckyj, J.C.; Taylor, K.D.; Coresh, J.; Rotter, J.I.; et al. Association of Mitochondrial DNA Copy Number With Cardiovascular Disease. JAMA Cardiol. 2017, 2, 1247–1255.
- Stewart, J.B.; Freyer, C.; Elson, J.L.; Larsson, N.G. Purifying selection of mtDNA and its implications for understanding evolution and mitochondrial disease. Nat. Rev. Genet. 2008, 9, 657–662.
- Sazonova, M.A.; Sinyov, V.V.; Barinova, V.A.; Ryzhkova, A.I.; Zhelankin, A.V.; Postnov, A.Y.; Sobenin, I.A.; Bobryshev, Y.V.; Orekhov, A.N. Mosaicism of mitochondrial genetic variation in atherosclerotic lesions of the human aorta. Biomed. Res. Int 2015, 2015, 825468.
- Sobenin, I.A.; Sazonova, M.A.; Postnov, A.Y.; Bobryshev, Y.V.; Orekhov, A.N. Changes of mitochondria in atherosclerosis: Possible determinant in the pathogenesis of the disease. Atherosclerosis 2013, 227, 283–288.
- Volobueva, A.; Grechko, A.; Yet, S.F.; Sobenin, I.; Orekhov, A. Changes in Mitochondrial Genome Associated with Predisposition to Atherosclerosis and Related Disease. Biomolecules 2019, 9, 377.
- Sazonova, M.A.; Sinyov, V.V.; Ryzhkova, A.I.; Galitsyna, E.V.; Khasanova, Z.B.; Postnov, A.Y.; Yarygina, E.I.; Orekhov, A.N.; Sobenin, I.A. Role of Mitochondrial Genome Mutations in Pathogenesis of Carotid Atherosclerosis. Oxid. Med. Cell Longev. 2017, 2017, 6934394.
- Sobenin, I.A.; Sazonova, M.A.; Postnov, A.Y.; Salonen, J.T.; Bobryshev, Y.V.; Orekhov, A.N. Association of mitochondrial genetic variation with carotid atherosclerosis. PLoS ONE 2013, 8, e68070.
- Sobenin, I.A.; Zhelankin, A.V.; Khasanova, Z.B.; Sinyov, V.V.; Medvedeva, L.V.; Sagaidak, M.O.; Makeev, V.J.; Kolmychkova, K.I.; Smirnova, A.S.; Sukhorukov, V.N.; et al. Heteroplasmic Variants of Mitochondrial DNA in Atherosclerotic Lesions of Human Aortic Intima. Biomolecules 2019, 9, 455.
- Dominic, E.A.; Ramezani, A.; Anker, S.D.; Verma, M.; Mehta, N.; Rao, M. Mitochondrial cytopathies and cardiovascular disease. Heart 2014, 100, 611–618.
- Beckman, K.B.; Ames, B.N. Oxidative decay of DNA. J. Biol. Chem. 1997, 272, 19633–19636.
- Yoo, S.M.; Jung, Y.K. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol. Cells 2018, 41, 18–26.
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and Mitophagy in Cardiovascular Disease. Circ. Res. 2017, 120, 1812–1824.
- Ko, M.S.; Yun, J.Y.; Baek, I.J.; Jang, J.E.; Hwang, J.J.; Lee, S.E.; Heo, S.H.; Bader, D.A.; Lee, C.H.; Han, J.; et al. Mitophagy deficiency increases NLRP3 to induce brown fat dysfunction in mice. Autophagy 2020, 1–17.
- Zhang, N.P.; Liu, X.J.; Xie, L.; Shen, X.Z.; Wu, J. Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. Lab. Invest. 2019, 99, 749–763.
- Orekhov, A.N.; Poznyak, A.V.; Sobenin, I.A.; Nikifirov, N.N.; Ivanova, E.A. Mitochondrion as a Selective Target for the Treatment of Atherosclerosis: Role of Mitochondrial DNA Mutations and Defective Mitophagy in the Pathogenesis of Atherosclerosis and Chronic Inflammation. Curr. Neuropharmacol. 2020, 18, 1064–1075.
- Orekhov, A.N.; Nikiforov, N.N.; Ivanova, E.A.; Sobenin, I.A. Possible Role of Mitochondrial DNA Mutations in Chronification of Inflammation: Focus on Atherosclerosis. J. Clin. Med. 2020, 9, 978.