G protein-coupled receptors (GPCRs) are cell surface receptors that respond to a wide variety of stimuli, from light, odorants, hormones, and neurotransmitters to proteins and extracellular calcium. GPCRs represent the largest family of signaling proteins targeted by many clinically used drugs.
- GPCR
- G protein
- GRK
- arrestin
- conformational change
- cell signaling
1. Introduction
G protein-coupled receptors (GPCRs) are the largest family of signaling proteins in animals. Among mammals, elephants hold the record with >3400 GPCR subtypes (Available online: http://sevens.cbrc.jp/). Humans express >800 different GPCRs, which are targeted by a greater number of clinically used drugs than any other protein family. Therefore, the molecular mechanisms of GPCR signaling have attracted close attention for several decades.
Rhodopsin, now considered a prototypical class A GPCR, was cloned before the idea that there is a class of rhodopsin-like receptors appeared, so it was compared to bacteriorhodopsin, a known protein with seven trans-membrane α-helices [1]. The cloning of the β2-adrenergic receptor a few years later revealed a similar topology [2], so 1986 should be considered as a year when the existence of a class of seven trans-membrane domain receptors, now called GPCRs or 7TMRs (seven trans-membrane domain receptors), was first demonstrated. The classification into five main families [3], which appears to hold water even today, brought certain order to the subsequent avalanche of GPCR sequences.
Rhodopsin also was the first GPCR, for which it was shown that activation involves a rigid body motion of trans-membrane α-helices [4] resulting in opening up of a cavity on the cytoplasmic side of the membrane. By that time, is was shown (again, in the visual system) that several proteins preferentially bind light-activated rhodopsin [5]. Thanks to numerous subsequent studies, today we know that three classes of proteins prefer active GPCRs over inactive: G proteins, G protein-coupled receptor kinases (GRKs), and arrestins (Figure 1). The latter preferentially bind to active phosphorylated receptors [6]. It appears that all three classes engage the cavity between the helices that opens on the cytoplasmic side of GPCRs upon receptor activation [7,8,9].
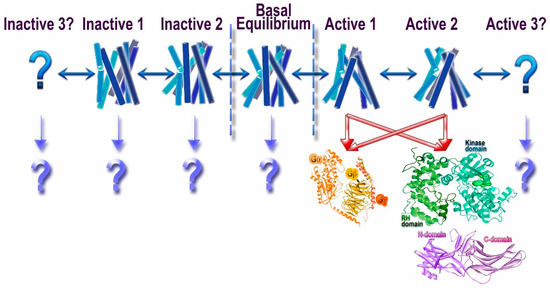
Thus, proteins belonging to these three families specifically engage activated GPCRs, which makes them candidate signal transducers. Classical view posits that GPCRs signal via G proteins (hence the name), whereas their phosphorylation by GRKs and subsequent arrestin binding serves to desensitize receptors [13]. This model implied that G proteins, GRKs, and arrestins recognize the same active GPCR conformation, in line with the idea that GPCRs exist in two distinct conformations, active and inactive [14]. However, biophysical studies suggested that GPCRs can exist in multiple conformations [10,15], including several distinct active conformations, depending on the ligand (Figure 1). The conformational equilibrium of GPCRs can never be shifted to a single inactive or active state, which likely explains documented constitutive activity of many receptors [10]. This complex behavior of GPCRs opened the possibility that different proteins interacting with activated GPCRs might prefer distinct receptor conformations. Indeed, quite a few ligands of several GPCRs were shown to preferentially activate G protein- or arrestin-mediated signaling, a phenomenon that was termed signaling bias [16] (Figure 1). It should be noted that the evolution apparently created GRKs and arrestins to suppress G protein-mediated signaling, so that in case of native ligands the families of conformations conducive to the binding of G proteins, arrestins, and GRKs largely overlap. However, even minor non-overlap can be exploited therapeutically by designing ligands pushing GPCRs into those rare conformations that are good enough for G protein, but not GRK or arrestin binding, or vice versa. The idea that certain GPCR conformations specifically enhance receptor interactions with some, but not all potential signal transducers appears very attractive. Indeed, synthetic agonists that facilitate G protein signaling but induce minimal desensitization (i.e., do not effectively enhance the binding of GRKs and/or arrestins) have been developed. There are fewer examples of ligands that promote arrestin binding but do not effectively induce G protein coupling. At the moment, we do not know which GPCR conformations favor which interaction partner, and how the structure of agonists needs to be changed to favor G proteins, as opposed to GRKs and/or arrestins. To complicate things further, it is conceivable that preferential engagement of particular signal transducers could be governed by the differences in conformational dynamics and/or kinetic rates between interchanging states rather than defined, distinct conformations. All of this also applies to the selection of a particular G protein in case of numerous GPCRs that couple to more than one subtype, or even to G proteins of different subfamilies.
2. GPCRs
Traditionally, it was assumed that GPCRs exist in two distinct conformations, active and inactive. This notion was the basis for the classical extended ternary complex model of GPCR-driven signaling [14]. This view implied that the active GPCR conformation preferred by G protein, GRKs, and arrestins is one and the same. However, biophysical experiments with purified fluorescently labeled β2-adrenergic receptor (β2AR) showed that the receptor can exist in multiple conformations, and that its conformational equilibrium is affected both by bound ligand and the presence of the cognate G protein [15]. The structure of agonist-liganded β2AR was puzzling: the receptor looked suspiciously similar to its structure with an inverse agonist carazolol [20]. Recent comprehensive biophysical study using both nuclear magnetic resonance (NMR) signals from 19F labeled Cys265 at the cytoplasmic end of trans-membrane helix 6 (TM6) and double electron-electron resonance (DEER) distance measurements between nitroxides introduced at position 148 at the cytoplasmic end of TM4 and 266 at the cytoplasmic end of TM6 revealed the complexity of the conformational equilibria of β2AR [10]. This study showed that ligand-free β2AR exists predominantly in two inactive conformations with rapid (hundreds of microseconds) transition between them. Even inverse agonists, while appreciably shifting the equilibrium, do not push the receptor into a single conformation. Interestingly, with bound agonist conformational heterogeneity increases, so that the receptor samples both inactive conformations, an intermediate one, and an active one, with the latter representing only a relatively small fraction of receptor population. The fully active conformation is stabilized only by G protein-mimicking single-chain camelid antibody (nanobody), but even then the heterogeneity persists [10]. These data explain the similarity of β2AR crystal structures with bound inverse agonist and high-affinity agonist. In addition, in agreement with these data, the structure of the same receptor with an agonist and bound nanobody stabilized the receptor in the high agonist affinity state, revealing much greater conformational changes on the cytoplasmic side than with agonist alone [21]. It is important to note that we do not currently have a full description of the conformational space explored by a GPCR under any condition: some states might be indistinguishable by NMR signal from 19F labeled Cys265, just like the intermediate and active states revealed by NMR were indistinguishable by the measurements of the distance between positions 148 and 266 using DEER [10]. Thus, any study, however comprehensive, can provide only a limited number of conformational states of the receptor, but not the full spectrum. An attractive idea of therapeutically exploitable biased signaling [22,23,24], implying that GPCRs can have distinct conformations preferred by G proteins, GRKs, and/or arrestins (Figure 1), is still awaiting experimental support.
The structure of the signaling β2AR complex with Gs (stabilized by a different nanobody that binds receptor-associated G protein) revealed an even larger movement of the cytoplasmic end of TM6 and identified receptor and G protein elements involved in their interaction [7]. Unexpectedly, this structure showed that only the Ras domain of the G protein α-subunit directly contacts the receptor [7]. Despite efforts of many labs, this breakthrough study remained the only GPCR-G protein complex structure available for several years. Based on this finding, another group designed a mini-G protein, essentially a mutated Ras domain of Gs α-subunit, and crystallized it with adenosine A2A receptor [25]. This work confirmed key findings of the original β2AR-Gs structure, thereby extending them to another class A receptor, although the use of engineered mini-G protein instead of a natural heterotrimer was a caveat. As crystallization of GPCR-G protein complexes remained tricky, two groups took advantage of improved cryo-electron microscopy (cryo-EM) technology to solve structures of two agonist-liganded class B receptors, calcitonin [26] and glucagon-like peptide-1 (GLP-1) [27], in complex with Gs. Both structures confirmed the previous idea that in class B GPCRs agonist peptides simultaneously bind to the large extracellular N-terminal domain and the pocket between helices occupied in class A GPCRs by their small molecule ligands. Both revealed significant conformational rearrangements of the extracellular part of the seven TM bundle to accommodate bound peptide, as well as a sharp kink in the TM6 necessary to ensure the outward movement of its cytoplasmic end to create a G protein-binding cavity.
Thus, at the moment, we have one atomic resolution crystal structure of heterotrimeric G protein with class A receptor, β2AR; a crystal structure of engineered mini-G protein with another class A receptor, adenosine A2A; and two lower resolution structures of active class B GPCRs in complex with G protein solved using cryo-EM. All structures show the interaction of the C-terminal α-helix of G protein α-subunit with the cavity that opens on the cytoplasmic side of GPCRs upon receptor activation and large outward movement of the cytoplasmic end of TM6 necessary to create this cavity. The main limitation of the available data is that in all cases the structures contain the same Gs protein (or its engineered mini-version), so that we cannot be sure that other classes of G proteins engage active GPCRs in the same way. Another limitation is that so far only one structure solved by X-ray crystallography contains natural heterotrimeric G protein. We certainly need structures with the highest possible resolution of GPCR complexes with other G proteins to draw general conclusions regarding GPCR-G protein interactions. These structures and/or biophysical studies would also show whether GPCRs of different classes undergo the same conformational changes upon activation.
Most importantly, we need unambiguous structural evidence for the basis of biased signaling, i.e., demonstration that distinct GPCR conformations are preferentially engaged by different signal transducers: G proteins, GRKs, and arrestins (Figure 1). In case of many GPCRs that couple to more than one kind of G proteins, it is tempting to think that the conformational requirements are also different. Thus far, these interesting ideas are largely speculative.
3. Signal Transducers
3.1. G Proteins
3.2. GRKs
3.3. Arrestins
3.3.1. Receptor Binding-Induced Structural Changes in Arrestins
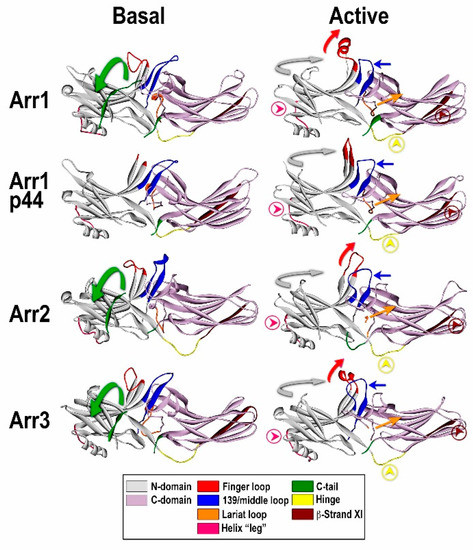
3.3.2. Receptor Specificity of Arrestins
3.3.3. Arrestin-Mediated Signaling
As GPCR-dependent arrestin-mediated signaling was recently covered in very comprehensive reviews [61,92,93], here we will focus on several examples of the rather unexpected GPCR-independent modes of arrestin signaling.
Among the four arrestin subtypes, only arrestin-3 facilitates JNK3 activation via the ASK1-MKK4/7-JNK3 cascade [94,95,96]. This function of arrestin-3 does not depend on its ability to bind receptors, as even receptor binding-deficient mutants facilitate JNK3 activation in cells [95,97]. One of the mutant forms of arrestin-3 was found to bind JNK3 and upstream kinases, but failed to promote JNK3 activation [97]. Importantly, this mutant was shown to act as a dominant-negative “silent scaffold”, recruiting the kinases away from productive scaffolds, thereby suppressing JNK3 activation in a dose-dependent manner [97]. This property made the mutant a potentially useful molecular tool for the suppression of often pro-apoptotic JNK3 activity, which might come in handy in cases where excessive cell death must be prevented, e.g., in degenerative disorders, such as Alzheimer’s or Parkinson’s disease.
Another cytoprotective tool with similar therapeutic potential, this time based on a different non-visual subtype, arrestin-2, emerged from the unexpected discovery of its role in the core mechanism of programmed cell death [98]. This study showed that caspase cleavage of arrestin-2 generates 1–380 fragment that cooperates with caspase-generated tBid to facilitate the release of cytochrome C from mitochondria, which is the point of no return in the apoptosis of vertebrate cells [99]. The expression of caspase-resistant form of arrestin-2, where the aspartates targeted by caspases were replaced with similarly negatively charged glutamates, which appears to retain other functions of the parental wild type protein, was shown to be cytoprotective.
The study of the arrestin-3 interaction with JNK3 identified three arrestin-3 elements involved in JNK3 binding, with the N-terminal 25-residue T1A peptide demonstrating the highest affinity for JNK3 [100]. Interestingly, the same short T1A peptide was found to interact with the upstream kinases and facilitate JNK3 activation in cells [101]. In addition to being the smallest MAP kinase scaffold known to date, this peptide, devoid of other arrestin-3 functions, appears to be a potentially useful tool for specific activation of pro-apoptotic signaling, which might provide novel therapeutic options in disorders associated with excessive cell proliferation, such as cancer. However, its ability to reduce cell growth and proliferation still needs to be tested.
Another unexpected finding was the elucidation of the role of arrestins in cell spreading and motility [102]. It was found that both non-visual subtypes play an important role in focal adhesion dynamics, recruiting clathrin to the microtubules that target focal adhesions, which apparently facilitates focal adhesion disassembly by promoting the internalization of integrins [102]. In the absence of both non-visual arrestins cells have too many abnormally long-lived focal adhesions. As the result, cells spread excessively and have trouble moving [102]. Interestingly, this arrestin function was also shown to be GPCR-independent.
Thus, arrestins not only mediate numerous signaling pathways initiated by activated GPCRs, but also participate in many receptor-independent signaling processes in the cell, affecting core cellular functions, such as adhesion, motility, survival, and apoptotic death. The structural basis of arrestin interactions with various non-receptor partners involved in GPCR-dependent and—independent arrestin signaling remains to be elucidated.
This entry is adapted from the peer-reviewed paper 10.3390/ijms18122519