2. Cardiomyocyte Embryonic Specification and Differentiation
The heart is the first functional organ to form during embryogenesis [
74]. Human cardiovascular system development begins during gastrulation at the end of the second week of gestation when the developing embryo’s metabolic demands exceed that supplied from placental diffusion alone [
75,
76]. Following gastrulation, the mammalian heart begins to develop from progenitor cells within the anterior lateral plate mesoderm [
77]. On approximately day 15 of gestation, the cardiac progenitor cells within the thoracic region condense into two bilaterally located endocardial tubes on either side of the trilaminar embryo.
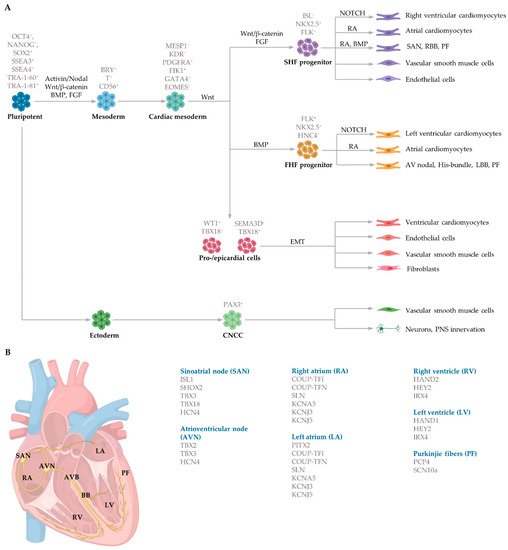
Mesoderm induction is regulated by several distinct and overlapping signalling pathways, including Activin and Nodal, both members of the Transforming growth factor beta (TGFβ) family, alongside the canonical (β-catenin-dependent) Wingless (Wnt) signalling pathway (A). Bone morphogenetic proteins (BMP), also members of the TGFβ family, and fibroblast growth factors (FGF) further aid in mesoderm determination in embryos [
78,
79]. The transcription factors homeobox protein NKX2.5 (NK2 transcription factor related, locus 5) and mesoderm posterior basic helix-loop-helix transcription factor 1 (MESP1) play a vital role in initiating cardiac commitment. NKX2.5 collaborates with the zinc finger protein transcription factors belonging to the GATA family to induce cardiomyocyte induction. Cardiac progenitors, cardiac mesoderm cells and cardiomyocytes in the left atria and ventricle express NKX2.5 [
80,
81]. Mesoderm induction commences in response to the secretion of growth factors from the adjacent endoderm germ layer. These factors include BMP4 and nodal signalling. The activation of the nodal signalling pathway in the proximal region of the primitive ectoderm initiates the expression of
Bmp4 within the adjacent extraembryonic ectoderm. BMP4 in the extraembryonic ectoderm then stimulates
Wnt3 expression in the epiblast, triggering the upregulation of mesoendodermal markers such as Brachyury (Bry) and Eomesodermin (EOMES), contributing to both the definitive endoderm and the cardiac mesoderm induction [
82]. EOMES-positive cells then activate MESP1 to control early cardiac progenitor commitment. MESP1 is the primary regulator of multipotent cardiac progenitor specification, regulating the induction of cardiac and mesoderm genes to activate the migration of mesodermal cells to the anterior lateral plate mesoderm via the primitive streak [
79,
83]. MESP1 activates Dickkopf Wnt signalling pathway inhibitor 1 (DKK1), thereby inhibiting Wnt and promoting cardiac differentiation and maturation [
84,
85]. Interestingly, Wnt/β-catenin signalling during cardiogenesis is biphasic. Prior to gastrulation, activation of Wnt/β-catenin signalling promotes cardiac lineage specification, whereas activation during gastrulation inhibits cardiomyocyte differentiation [
86,
87].
During cardiac crescent formation, two distinguishable cell populations exist: cells within the splanchnic mesoderm and cells in the epithelial structure of the crescent [
77]. These different populations of progenitor cells, referred to as the first heart field (FHF) and second heart field (SHF), produce different regions of the developing heart. MESP1 controls both the distinct populations of the FHF and SHF during early gastrulation which give rise to the precursors of the subtype-specific cardiomyocytes. Early cardiac progenitors, positive for the marker ISL1, induce the production of FHF and SHF progenitor cells. FHF progenitors (ISL1- NKX2.5+), which control myocardium specification, are regulated by BMP and contribute to portions of the atria and left ventricle and the atrioventricular bundle conduction cells [
88]. The SHF progenitors (ISL1 + NKX2.5+) that regulate both myocardium and endocardium specification are controlled by FGF and WNT and give rise to portions of the atria, right ventricle and SAN conduction cells [
89,
90] (A).
Cardiac mesoderm progenitors coalesce, forming two paired primordia heart tubes that are composed of both endocardial and myocardial lineage. Subsequently, FHF- and SHF-derived cardiac progenitor cells proliferate within the developing structure [
91]. At approximately day 21 of gestation, the bilaterally paired tubes fuse, forming a primitive linear tube. Blood flows into the tube at the caudal side and outflows at the cranial side [
74]. During early heart development, dominant pacemaker activity arises at the intake region at the caudal end of the primitive tube, referred to as the sinus venosus. The embryonic cells within the sinus venosus are characterised by the expression of HCN4, encoding the potassium/sodium hyperpolarized-activated cyclic nucleotide-gated channel 4 responsible for the hyperpolarisation-activated current essential for pacemaking ability [
92]. The heart tube is elongated as FHF- and SHF-derived progenitor cardiac cells rapidly proliferate and differentiate into cardiomyocytes in response to NOTCH and retinoic acid signalling from the endocardium and epicardium, respectively. Retinoic acid, the active derivative of vitamin A, modifies the expression of critical genes to induce cardiomyocyte differentiation by the induction of fibroblast growth factor signalling [
93]. NOTCH coordinates cardiac specification by interaction with other signalling pathways, including WNT and BMP [
94].
3. Generation of Subtype-Specific Cardiomyocytes from Human Pluripotent Stem Cells
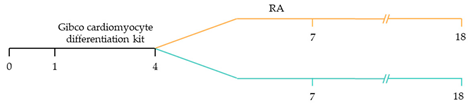
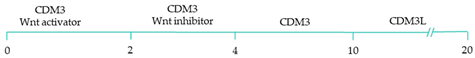
There are a variety of protocols enabling the generation of human cardiomyocytes from hiPSCs [
95]. Such approaches include the use of growth factors, small molecules, genetic manipulation and biophysical cues within both monolayer and three-dimensional (3D) cultures [
96]. However, these studies typically yield mixed populations of atrial, ventricular and pacemaker-like cardiomyocytes [
97,
98,
99]. The mixed subtypes of cardiomyocytes may distort innate cellular physiology, confound disease phenotypes and jeopardize the therapeutic effects of transplanted cardiomyocytes for cardiac tissue regeneration [
92]. Accordingly, at the forefront of cardiac engineering is the ability to generate subtype-specific cardiomyocytes for the precise fabrication of atrial, ventricular and pacemaker-like human-induced pluripotent stem cell (hiPSC)-derived cardiomyocytes. Recent research has identified that subtype-specific cardiomyocytes from hiPSCs can be generated by imitating embryonic heart chamber development [
100,
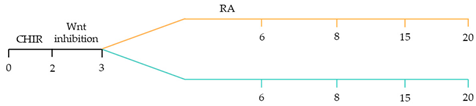
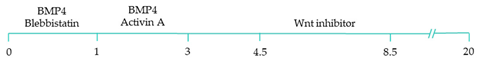
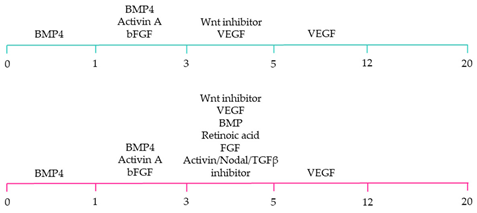
101]. These strategies have focused on the manipulation of the critical developmental signalling pathways of cardiogenesis, in particular, WNT, NOTCH and retinoic acid signalling pathways () [
22].
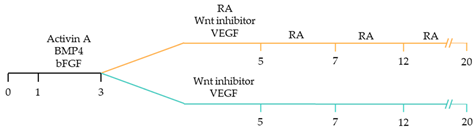
NOTCH signaling is vital for ventricular cardiomyocyte specification and morphogenesis. Moreover, NOTCH regulates coronary vessel specification and is a primary modulator of arteriovenous specification during vessel development [
102]. The NOTCH signalling pathway consists of two families of membrane-bound ligands, Jagged1 and 2, and Delta-like (DLL)1, 3 and 4, that bind to four transmembrane-receptors, NOTCH 1 to 4, of an adjacent cell [
103]. Ligand interaction with these receptors initiates a cascade of proteolytic cleavages, including the proteolytic cleavage-by-γ-secretase leading to the NOTCH intracellular domain release from the cell-membrane and subsequent translocation to the nucleus [
104]. In the nucleus, the NOTCH intracellular domain reacts with the recombining binding protein suppressor of hairless protein, resulting in the expulsion of a histone deacetylase corepressor complex, thus giving rise to an activation complex [
105]. Further, Mastermind-like protein 1 and histone acetyltransferase are recruited to the site, activating the transcription of NOTCH target genes, such as the basic helix–loop–helix transcriptional repressors
HEY1 (Hairy/enhancer-of-split related with YRPW motif protein 1) and
HEY2 (Hairy/enhancer-of-split related with YRPW motif protein 2).
The activation of these NOTCH target genes results in ventricular chamber development and spatial arrangement of cardiomyocytes within the myocardium [
94].
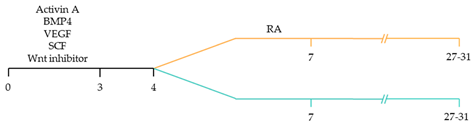
Recent studies have demonstrated that retinoic acid signalling can promote atrial-specific differentiation of hiPSCs through the upregulation of the transcription factor COUP-TFII [
106]. Conversely, the lack of retinoic acid is associated with ventricular-specific differentiation [
100]. In particular, retinoic acid aids in atrial/ventricular-specific hiPSC differentiation during a restrictive temporal window proceeding mesoderm commitment [
22,
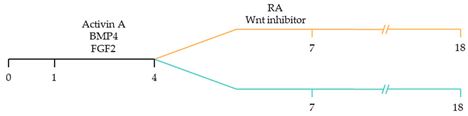
23]. Moreover, retinoic acid-mediated chamber specific differentiation of hiPSCs depends on the distinct mesoderm progenitor population. Different concentrations of BMP4 and Activin A induce two mesoderm populations characterised by unique cell surface markers, i.e., CD235a+ CYP26A1+ and RALDH2+ ALDH+, which generate ventricular and atrial cardiomyocytes, respectively. During hiPSC differentiation, retinoic acid signalling will efficiently give rise to atrial cardiomyocytes in RALDH2+ ALDH+ mesoderm progenitors. Comparatively, a lack of retinoic acid signalling will generate ventricular cardiomyocytes in CD235a+ CYP26A1+ progenitors [
22]. Further, retinoic acid signalling combined with WNT and BMP guides the differentiation of vascular smooth muscle and cardiac fibroblast lineage from hiPSCs. Moreover, the combined treatment of RA and BMP signalling directs hiPSC differentiation towards pacemaker-like cardiomyocytes [
107]. Interestingly, the inhibition of COUP-TFII signalling ablates the repressing effect on NOTCH signalling, thereby indirectly promoting ventricular differentiation [
106]. Inhibition of retinoic acid signalling indirectly promotes cardiac progenitors toward a ventricular-like subtype [
100]. Additionally, combined treatment with BMP4, Activin A and the WNT signalling pathway inhibitor is highly efficient at stimulating hiPSC ventricular subtype specification in differentiating cardiomyocytes [
108].
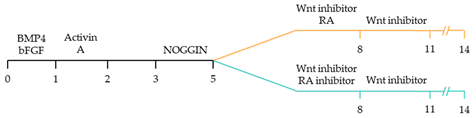
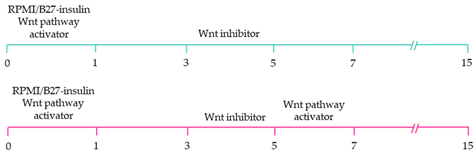
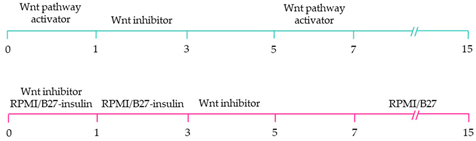
The production of pacemaker-like cardiomyocytes from hiPSCs has also been achieved by mirroring embryonic pacemaker lineage specification. Sinoatrial cells originate from TBX18+ NKX2.5− mesoderm progenitors, distinguishing them from chamber-specific cardiomyocytes and other components of the CCS [
109]. Pacemaker-like cardiomyocyte induction using transient stage-specific exposure to FGF and BMP signalling with concomitant inhibition of Wnt signalling produced relatively pure populations of pacemaker-like cardiomyocytes [
110]. However, more recently, Ren, et al. [
111] demonstrated that canonical Wnt signalling directs pacemaker-like specification of differentiating hiPSC-derived cardiomyocytes. Wnt signalling promoted enhanced expression of pacemaker-specific genes such as SHOX2, HCN4, ISL1, TBX3 and TBX18 with a concomitant reduction in NKX2.5 expression. Wnt-activated cardiomyocytes demonstrated less negative resting membrane potentials corresponding with native pacemaker electrophysiological properties, thereby functionally supporting genetic alterations. Moreover, the Wnt-activated hiPSC-derived pacemaker-like cardiomyocytes augmented pacing of bioengineered “mini heart” models. Similarly, Protze, Liu, Nussinovitch, Ohana, Backx, Gepstein and Keller [
107] also used a transgene-independent method to produce pacemaker-like cardiomyocytes from hiPSCs and human embryonic stem cells by stage-specific alteration of developmental signal pathways. More specifically, FGF signalling was inhibited at the developmental stage corresponding to cardiac mesoderm induction, and BMP and retinoic acid signalling pathways were activated. Hence, the development of NKX2.5+ cardiomyocytes was eradicated, thereby directing cardiomyocyte differentiation towards a pacemaker specific lineage. Electrophysiological analysis demonstrated that the majority (90%) of pacemaker-specific cardiomyocytes had characteristic pacemaker action potentials representative of native pacemaker cardiomyocytes, suggesting typical pacemaker ion current profiles. Remarkably, when transplanted into rat hearts, the bioengineered cardiomyocytes functioned as biological pacemakers, capable of altering the pace of the host heart. Transgenic approaches to generate pacemaker-like cardiomyocytes from hiPSCs include the forced expression of SHOX2, TBX3 and TGF-beta activated kinase [
112,
113,
114].
Alternatively, the native cellular interactions can be recapitulated in vivo to aid subtype-specific differentiation to provide an alternative protocol to achieve pacemaker-like specific cardiomyocytes. Schweizer, Darche, Ullrich, Geschwill, Greber, Rivinius, Seyler, Müller-Decker, Draguhn, Utikal, Koenen, Katus and Thomas [
52] generated hiPSC pacemaker-specific cardiomyocytes by initially co-culturing hiPSCs with visceral endoderm-like (END-2) cells for ten to twelve days to form beating cell aggregates. The beating cell aggregates were then isolated and transferred to foetal bovine serum-enriched medium for further culturing for six weeks. Interestingly, the expressions of HCN1, HNC4, NCX1, Ca
v1.2 and Ca
v1.3 within the beating clusters were similar to, or higher than those of human sinoatrial nodal cells. Action potential measurements of cells isolated from clusters demonstrated predominantly nodal-type (63.4%) with less atrial-type (36.6%) action potential configurations and no ventricular action potential morphologies. However, the relative expression of connexin demonstrated enhanced pacemaker lineage induction, as demonstrated by a fourfold increase in Cx45 levels following differentiation. Furthermore, the beating clusters demonstrated constant and synchronised pacing of neonatal rat ventricular myocytes in co-culture experiments.
Whilst these findings aid in generating subtype-specific cardiomyocytes derived from hiPSCs, the clinical application of these methods remain limited as they yield mixed cardiomyocyte subtype-populations. Moreover, the aforementioned protocols produce cardiomyoctes with relatively immature structure and function than their native counterparts. Further refinement of the generation of subtype-specific cardiomyocytes promises to propel the translational applications of patient-derived hiPSCs for disease modelling, drug discovery/toxicity and regenerative medicine.