Autophagy is a conserved cell quality control system, and increasing evidence suggests that it plays an important role in numerous and different biological processes, such as starvation, aging, inflammation, and organ remodeling, by maintaining cellular homeostasis.
- malnutrition
- autophagy
- heart failure
- chronic diseases
- amino acids
1. Introduction
Autophagy occurs through the removal and degradation of intracellular components to recycle molecules needed for the synthesis of others. Three main types of autophagy have been identified: (1) macro-autophagy (commonly named autophagy), (2) micro-autophagy, and (3) chaperone-mediated autophagy. Macro-autophagy occurs by vesicle formation (autophagosome) to incorporate portions of the cytoplasm that then merge with lysosomes and result in digestion of the contents [1][2]. The presence of autophagy is demonstrated by the expression of autophagy-related proteins, such as light chain 3 (LC3), Beclin-1, autophagy-related gene (ATG), 5–12 complex, and p62 [3]. Micro-autophagy is also a degradation system that takes place directly on the surface of the lysosomes [4][5]. Finally, chaperone-mediated autophagy allows the selective degradation of only soluble proteins [2]. The prevalent function of autophagy is to promote cell survival in stressful conditions like starvation and preserve energy status in response to energy deprivation, hypoxic conditions, and high temperatures through digesting cellular components and recycling of essential elements for reuse [6][7]. Altered autophagy is also associated with many pathological states such as cancer, neurodegenerative disorders, myopathies, and cardiomyopathies [8].
2. Autophagy in the Heart
Autophagy in the heart occurs naturally at basal levels to maintain normal physiological cell functions [9]. However, it can be intensively activated in response to stressful situations such as decreased energy under the form of adenosine triphosphate (ATP) and increased oxidative stress, thus playing a pro-survival role [10]. During aging, although the rate of autophagosome formation and the efficiency of autophagosome-lysosome fusion as well as the proteolytic activity of lysosomes decline with age, continuous removal of exhausted or damaged components by efficient autophagy machinery, and their replacement with newly synthesized molecules, ensures cellular homeostasis and delays the aging process [10]. Indeed, it has been shown that lifelong caloric restriction by 40% increases the expression of autophagic markers in the heart [11]. The increase in autophagy resulting from caloric restriction may have a protective role on the cardiomyocytes by reducing levels of oxidative damage due to aging and cardiovascular diseases [12]. On the contrary, over-activated autophagy may deplete molecules and organelles fundamental for cellular survival, thus driving cells to death [13][14]. Autophagic degradation of “self” proteins for the production of AAs is also important for survival during neonatal starvation [15]. A state of nutrient deprivation, due to starvation or myocardial ischemia, induces autophagy, generating fatty acids and AAs that enter the Krebs cycle generating ATP and promoting survival of cardiac cells [16][17]. Although evidence indicates that physiological autophagy plays important roles in maintaining heart homeostasis, the excess of autophagy could favor and exacerbate heart-related diseases.
3. Autophagy in Heart Failure
Autophagy has been reported to play a role in the pathophysiology of human heart failure [8][10][18]. Many forms of heart failure are associated with the accumulation of misfolded proteins due to their impaired degradation and, hence, autophagy is fundamental under stress conditions like ischemia, starvation, and β-adrenergic stimulation, where it acts as a pro-survival mechanism by removing misfolded or anomalous proteins and damaged cellular structures [19].
Brief periods of ischemia may induce heart autophagy. In mouse hearts in vivo [16] and isolated rabbit hearts [20], autophagy was induced by ischemia and further enhanced by reperfusion. In hypoxic and re-perfused rabbit hearts, autophagosome formation has been observed after 20–40 min [21], and this is associated with functional myocyte recovery. More recently, it has been suggested that autophagy also enhances the survival of cardiomyocytes in hearts exposed to permanent coronary artery occlusion [22]. However, although many studies showed that induction of autophagy could preserve heart function during ischemia/reperfusion injury, others have suggested that autophagy contributes to cell death [3]. The magnitude of autophagy within the cell may contribute to its protective or detrimental role. Indeed, in human warm blood, cardioplegic arrest caused myocyte autophagy, with a magnitude and severity that were proportional to the length of cardioplegic arrest [23].
Autophagy has been recently extensively detected in patients with end-stage heart failure and precedes and sets the stage for the occurrence of apoptosis, oncosis, and necroptosis, which only rarely start independently of autophagy. The autophagic process progresses with broad cytosolic destruction and nuclear disintegration, finally resulting in cell death by necroptosis and, to a lesser extent, by apoptosis [24]. Then, autophagy seems to be a primary driving force leading to the progressive cardiomyocyte cell loss observed in end-stage heart failure [24]. It is interesting to note that the nucleus is the early autophagic target, and it develops typical erosions as if it had been caused by a bite (Figure 1A–C).
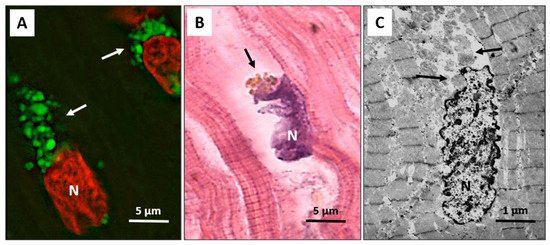
Figure 1. Failing heart. (A) Typical anti-LC3 immunofluorescence (green) of cardiomyocytes in an advanced stage of autophagic damage. Green bodies (arrows) are present around and much more at the pole of nuclei. Cardiomyocytes with massive green staining, in combination with the red staining of the nucleus, assume a “strawberry-like” appearance. N = nucleus in red; (B) Damaged cardiomyocytes show nuclei (N) very irregular in shape as if he had been bitten. Around these nuclei, the cytoplasm is devoid of organelles, and there are always clumps of brown debris bodies (arrow) (eosin and hematoxylin staining); (C) Representative transmission electron microscopy pictures show a damaged cardiomyocyte, with chromatin condensation in the nucleus (N) and deep invaginations of the nuclear envelope. Cytoplasmic granular material without organelles is present around the nucleus and near the nuclear pole (arrows).
In advanced stages of the autophagic process, the LC3-positive vacuoles develop TUNEL positivity, indicating that DNA fragments from the nucleus have been encapsulated within autophagosomes. These data are consistent with the possibility that autophagy is a primary driving force leading to progressive cardiac cell loss in end-stage heart failure [24]. The link between CD and CHF, malnutrition, HCS, and autophagy is schematized in Figure 2.
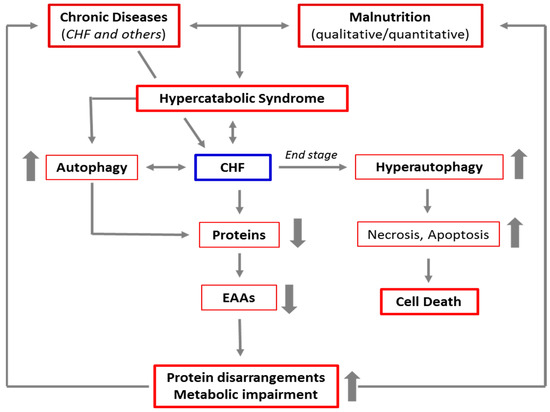
Figure 2. Schematic representation of the link between chronic diseases, malnutrition, hypercatabolic syndrome, and autophagy. The increase in catabolism induced by chronic diseases and malnutrition favors the activation of autophagy to guarantee the cells sufficient energy and materials to cope with accelerated metabolism. This results in a cascade of events, leading to protein disarrangements (the severe alteration of the protein components of the body), and then to metabolic impairment. This creates a vicious circle which, in the absence of adequate nutritional interventions, maintains and favors the hypercatabolic syndrome. Furthermore, in patients with advanced chronic diseases, like the end stage of CHF, autophagy is severely triggered, and the autophagic machinery may also drive the cells to self-destruction.
In specific conditions, excessive activation of autophagy can lead to the destruction of essential molecules and organelles favoring cell death [12][14] (autophagy-dependent cell death) with no signs of apoptosis or necrosis. This situation was recently named autosis [25].
4. Role of Mammalian Target of Rapamycin (mTOR) in Autophagy
Autophagy is a dynamic process that depends on strict coordination and regulation of multiple enzymatic pathways as Beclin-1/class III phosphatidylinositol-3 kinase (PI-3K), PI-3K/Akt/mTOR pathways, and AMPK/mTOR. The last is directly influenced by the availability of nutrients, AAs in particular [26].
Usually, autophagy starts under conditions of energy deprivation in response to starvation with reduction of ATP synthesis. Adenosine monophosphate (AMP)-activated protein kinase (AMPK), an energy-sensing kinase, is activated when the concentration of ATP decreases. Under starvation (such as in ischemia or malnutrition), AMPK acts as a checkpoint by suppressing cellular growth and by promoting the activation of autophagy in cardiomyocytes. In fact, inhibition of AMPK reduces autophagy and increases cardiomyocyte cell death [17]. AMPK acts through ULK1 (Unc-51 like autophagy activating kinase) activation. In fact, ULK1 is a serine/threonine-protein kinase that mediates the induction of autophagy [27]. On the contrary, mTOR modulates autophagy by inhibiting the ULK1-kinase; it counteracts the activation of autophagy induced by AMPK. Thus, the AMPK-mTOR axis is crucial for the control of autophagy during energy stress and starvation [12]. On this basis, the excess of AMPK-induced autophagy, in a state of chronic disease or malnutrition, may be controlled by modulating mTOR activity.
mTOR is a highly conserved serine-threonine kinase that belongs to the PIKK (phosphoinositide 3-kinase related protein kinase) superfamily, which includes several kinases involved in nutrient sensing and DNA repair [28][29][30]. mTOR represents the crossroad of numerous biochemical pathways with functions that can be very different, sometimes even opposing each other. Its action, sometimes paradoxical, could be due to the fact that mTOR is the catalytic subunit of two distinct complexes: mTORC1 and mTORC2. mTORC1, localized on the outer membrane of lysosomes, and mTORC2, whose function requires association with ribosomes [31]. mTOR is activated by different stimuli, such as nutrients (nitrogen substrates provided by digestion of proteins or AAs), growth factors, energy and stress signals, and exercise. In response to these stimuli, mTORC1 mediates cell growth and proliferation. Then, mTOR acts as the center of a complex pathway network that controls protein synthesis, cell differentiation, growth, and proliferation [32][33][34]. In addition, energy variations and glucose availability are sensed by AMPK, which works coordinately with mTORC1 by shifting the cells to catabolic metabolism [35]. However, when AA availability is limited, mTOR can also operate through mTORC2 to promote autophagy [36]. Under these conditions, the role of mTORC1 shifts from suppressor to activator of autophagy, and the reactivation of mTOR is dependent on AAs one of the end products of autolysosomal degradation [37]. The different degrees of modulation of the mTORC1/mTORC2 complexes could shift a cell’s fate from survival to death and vice-versa.
It has been shown that AAs and insulin stimulate translational control of protein synthesis but in different ways. Indeed, AAs do not activate phosphoinositide-3-kinase and protein kinase B (Akt) but stimulate mTOR indirectly through TSC1/2 (tuberous sclerosis 1 and 2 alias hamartin) downregulation and Rheb (Ras homolog enriched in brain) activation in skeletal muscle of aged animals [38]. It is interesting to note that mTOR is downregulated by treatment with AAs in elderly rat hearts [26]. AAs, as well as other nutrients and physical exercise, maintain anabolism by stimulating protein synthesis in skeletal and cardiac muscles through the phosphorylation of the ribosome-associated S6 kinase. This S6-kinase activation favors a high level of translation of mRNAs that encode ribosomal proteins, thus activating both cell entry of AAs and protein synthesis [39]. In addition, AAs repress autophagy by activating the mTOR (mTORC1-mediated) metabolic pathway [40][26][41]. Indeed, AAs are essential for mTORC1 activation. When there is nutrient availability, mTOR negatively regulates autophagy via mTORC1. On the contrary, in the setting of a deficiency of AAs, mTOR is not efficiently activated by other stimuli [42][43] and can promote autophagy by operating through the mTORC2 complex [36]. The influence of AAs/nutrition, ATP/AMP, starvation, and caloric restriction on mTORC1 and autophagy is schematized in Figure 3.
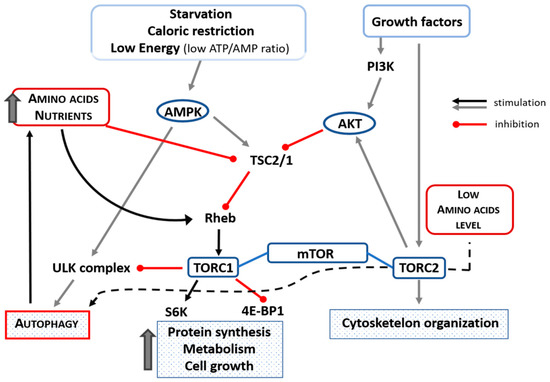
Figure 3. Schematic representation of the influence of amino acids, nutrients, starvation, low ATP/AMP ratio, and caloric restriction on mTORC1 and autophagy. Amino acid availability inhibits autophagy by inhibiting TSC2/1 and activating Rehb that could facilitate the transport of amino acids into the cell and which, in turn, could activate TORC1 that inhibits ULK-complex (black line). In case of low availability of amino acids, autophagy is activated through the TORC2 complex (black dotted line). Up thick arrow = increase. Akt = protein kinase B; ATP = adenosine triphosphate; 4EBP1 = eIF4E-binding protein-1; PI3K = phosphoinositide 3-kinase; Rheb = Ras homolog enriched in brain; S6 = S6-kinase; TSC1/2 = tuberous sclerosis complex 1/2; ULK (Unc-51 like autophagy activating kinase).
This entry is adapted from the peer-reviewed paper 10.3390/ijms22073332
References
- Shibutani, S.T.; Saitoh, T.; Nowag, H.; Münz, C.; Yoshimori, T. Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 2015, 16, 1014–1024.
- Cuervo, A.M.; Macian, F. Autophagy, nutrition and immunology. Mol. Asp. Med. 2012, 33, 2–13.
- Lin, X.; Xiao, W.; Xiao, L.; Liu, M. Molecular mechanisms of autophagy in cardiac ischemia/reperfusion injury (Review). Mol. Med. Rep. 2018, 18, 675–683.
- Li, W.-W.; Li, J.; Bao, J.-K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136.
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41.
- Tsujimoto, Y.; Shimizu, S. Another way to die: Autophagic programmed cell death. Cell Death Differ. 2005, 12, 1528–1534.
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616.
- Kostin, S. Pathways of myocyte death: Implications for development of clinical laboratory biomarkers. Adv. Clin. Chem. 2005, 40, 37–98.
- Chiong, M.; Wang, Z.V.; Pedrozo, Z.; Cao, D.J.; Troncoso, R.E.; Ibacache, M.; Criollo, A.; Nemchenko, A.A.; Hill, J.; Lavandero, S. Cardiomyocyte death: Mechanisms and translational implications. Cell Death Dis. 2011, 2, e244.
- De Meyer, G.R.Y.; De Keulenaer, G.W.; Martinet, W. Role of autophagy in heart failure associated with aging. Heart Fail. Rev. 2010, 15, 423–430.
- Wohlgemuth, S.E.; Julian, D.; Akin, D.E.; Fried, J.; Toscano, K.; Leeuwenburgh, C.; Dunn, J.W.A. Autophagy in the Heart and Liver During Normal Aging and Calorie Restriction. Rejuvenation Res. 2007, 10, 281–292.
- Ahn, J.; Kim, J. Nutritional Status and Cardiac Autophagy. Diabetes Metab. J. 2013, 37, 30–35.
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752.
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010.
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nat. Cell Biol. 2004, 432, 1032–1036.
- Matsui, Y.; Takagi, H.; Qu, X.; Abdellatif, M.; Sakoda, H.; Asano, T.; Sadoshima, J. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 2007, 100, 914–922.
- Loos, B.; Lochner, A.; Engelbrecht, A.M. Autophagy in heart disease: A strong hypothesis for an untouched metabolic reserve. Med. Hypotheses 2011, 77, 52–57.
- Hein, S.; Arnon, E.; Kostin, S.; Schönburg, M.; Elsässer, A.; Polyakova, V.; Schaper, J. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: Structural deterioration and compensatory mechanisms. Circulation 2003, 107, 984–991.
- Ghosh, R.; Pattison, J.S. Macroautophagy and Chaperone-Mediated Autophagy in Heart Failure: The Known and the Unknown. Oxid. Med. Cell. Longev. 2018, 2018, 8602041.
- Decker, R.S.; Wildenthal, K. Lysosomal alterations in hypoxic and reoxygenated hearts. I. Ultrastructural and cytochemical changes. Am. J. Pathol. 1980, 98, 425–444.
- Liu, X.; Van Vleet, T.; Schnellmann, R.G. The role of calpain in oncotic cell death. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 349–370.
- Zhang, Z.; Li, H.; Chen, S.; Li, Y.; Cui, Z.; Ma, J. Knockdown of microRNA-122 protects H9c2 cardiomyocytes from hypoxia-induced apoptosis and promotes autophagy. Med. Sci. Monit. 2017, 23, 4284–4290.
- Chen-Scarabelli, C.; Faggian, G.; Shah, M.; Saravolatz, L., II; Saravolatz, S.; Scarabelli, G.; Scarabelli, T.M. Warm blood cardioplegia induces myocyte autophagy, whose magnitude and severity are proportional to the duration of cardioplegic arrest. Circulation 2010, 122, A142.
- Corsetti, G.; Chen-Scarabelli, C.; Romano, C.; Pasini, E.; Dioguardi, F.S.; Onorati, F.; Knight, R.; Patel, H.; Saravolatz, L.; Faggian, G.; et al. Autophagy and Oncosis/Necroptosis Are Enhanced in Cardiomyocytes from Heart Failure Patients. Med. Sci. Monit. Basic Res. 2019, 25, 33–44.
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M.; Wei, Y.; Ginet, V.; Zhang, L.A.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+, K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371.
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141.
- Morita, M.; Gravel, S.-P.; Hulea, L.; Larsson, O.; Pollak, M.; St-Pierre, J.; Topisirovic, I. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle 2015, 14, 473–480.
- Albert, V.; Hall, M.N. mTOR signaling in cellular and organismal energetics. Curr. Opin. Cell Biol. 2015, 33, 55–66.
- Howell, J.J.; Manning, B.D. mTOR couples cellular nutrient sensing to organismal metabolic homeostasis. Trends Endocrinol. Metab. 2011, 22, 94–102.
- Flati, V.; Corsetti, G.; Pasini, E.; Rufo, A.; Romano, C.; Dioguardi, F.S. Nutrition, Nitrogen Requirements, Exercise and Chemotherapy-Induced Toxicity in Cancer Patients. A Puzzle of Contrasting Truths? Anti Cancer Agents Med. Chem. 2015, 16, 89–100.
- Nijhout, H.F.; Callier, V. A new mathematical approach for qualitative modeling of the insulin-TOR-MAPK network. Front. Physiol. 2013, 4, 245.
- Evans, D.S.; Kapahi, P.; Hsueh, W.-C.; Kockel, L. TOR signaling never gets old: Aging, longevity and TORC1 activity. Ageing Res. Rev. 2011, 10, 225–237.
- Navé, B.T.; Ouwens, M.; Withers, D.J.; Alessi, D.R.; Shepherd, P.R. Mammalian target of rapamycin is a direct target for protein kinase B: Identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem. J. 1999, 344, 427–431.
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023.
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946.
- Deldicque, L.; Theisen, D.; Francaux, M. Regulation of mTOR by amino acids and resistance exercise in skeletal muscle. Graefes Arch. Clin. Exp. Ophthalmol. 2005, 94, 1–10.
- Proud, C.G. mTOR-mediated regulation of translation factors by amino acids. Biochem. Biophys. Res. Commun. 2004, 313, 429–436.
- Flati, V.; Pasini, E.; D’Antona, G.; Speca, S.; Toniato, E.; Martinotti, S. Intracellular Mechanisms of Metabolism Regulation: The Role of Signaling via the Mammalian Target of Rapamycin Pathway and Other Routes. Am. J. Cardiol. 2008, 101, S16–S21.
- Hara, K.; Yonezawa, K.; Weng, Q.-P.; Kozlowski, M.T.; Belham, C.; Avruch, J. Amino Acid Sufficiency and mTOR Regulate p70 S6 Kinase and eIF-4E BP1 through a Common Effector Mechanism. J. Biol. Chem. 1998, 273, 14484–14494.
- D’Antona, G.; Ragni, M.; Cardile, A.; Tedesco, L.; Dossena, M.; Bruttini, F.; Caliaro, F.; Corsetti, G.; Bottinelli, R.; Carruba, M.O.; et al. Branched-Chain Amino Acid Supplementation Promotes Survival and Supports Cardiac and Skeletal Muscle Mitochondrial Biogenesis in Middle-Aged Mice. Cell Metab. 2010, 12, 362–372.
- Wang, X.; Campbell, L.E.; Miller, C.M.; Proud, C.G. Amino acid availability regulates p70 S6 kinase and multiple translation factors. Biochem. J. 1998, 334, 261–267.
- Volpi, E.; Campbell, W.W.; Dwyer, J.T.; Johnson, M.A.; Jensen, G.L.; Morley, J.E.; Wolfe, R.R. Is the optimal level of protein intake for older adults greater than the rec-ommended dietary allowance? J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 677–681.
- Morley, J.E.; Argiles, J.M.; Evans, W.J.; Bhasin, S.; Cella, D.; Deutz, N.E.; Doehner, W.; Fearon, K.C.; Ferrucci, L.; Hellerstein, M.K.; et al. Nutritional Recommendations for the Management of Sarcopenia. J. Am. Med. Dir. Assoc. 2010, 11, 391–396.