γ-Glutamyltransferase (GGT), a membrane-bound enzyme, contributes to the metabolism of glutathione (GSH), which plays a critical physiological role in protecting cells against oxidative stress. GGT has been proposed as a biomarker of carcinogenesis and tumor progression given that GGT activity is important during both the promotion and invasion phases in cancer cells.
- biomarkers
- γ-glutamyltransferase
- glutathione
- prognosis
- urologic neoplasms
1. Introduction
γ-Glutamyltransferase (GGT) is one of the enzymes involved in the γ-glutamyl cycle, which contributes to the synthesis and degradation of glutathione (GSH) [1]. GSH is known to be a major intracellular water-soluble antioxidant playing a protective role against reactive oxygen species (ROS) [2]. GGT also has a possible, albeit controversial, role in the transportation of amino acids across cellular membranes [3]. In addition to being used as a hepatobiliary biomarker that is especially sensitive to excessive alcohol consumption [4], elevated serum GGT has been shown to be an adverse prognostic factor in patients with life-threatening diseases. A recent study reported on the utility of the serum GGT level as a predictive biomarker of clinical outcomes in patients with a multitude of conditions, including cardiovascular diseases, diabetes mellitus, and metabolic syndrome [4]. Moreover, serum GGT is reportedly associated with a poor prognosis in patients with liver cancer, reflecting unfavorable clinicopathological features, including vascular invasion and the tumor burden, as demonstrated in previous meta-analyses [5][6]. Subsequently, serum GGT was also found to be a significant prognostic biomarker in patients with a variety of malignant neoplasms; however, reports of serum GGT in relation to genitourinary (GU) cancer are relatively limited, and the findings among studies of renal cell carcinoma (RCC) are sometimes contradictory depending on the metastatic status of the disease [7][8][9][10][11][12]. On the other hand, serum GGT was found to be significantly and independently associated with shorter overall survival in patients with advanced GU cancer, such as metastatic prostate cancer (PC) and advanced urothelial carcinoma (UC) [13][14].
2. Structure, Functions, and Expression of GGT
2.1. GGT Family Enzymes and Structure
There are at least eight potential full-length GGT family proteins, including GGT1, GGT2, GGT3P, GGT4P, GGT5, GGT6, GGT7, and GGT8P [15]. Of these, GGT1 and GGT5 are the only two enzymes that have been shown to be catalytically active [15][16]. Immunolabelling of these closely related enzymes in human tissues has revealed different expression patterns in different organs or even within the same tissue (e.g., GGT1 is expressed on the apical surface of the renal proximal tubules whereas GGT5 is expressed in the interstitial cells of the kidney), resulting in access to different substrates [17]. Moreover, their enzymatic activities are not at the same level according to a previous kinetic analysis, which demonstrated that GGT1 is able to cleave GSH approximately 46 times faster than GGT5 [18]. Furthermore, a genetic deficiency of the GGT1 gene was found to cause severe growth failure or skeletal abnormalities while null mutants of GGT5 gene have not been associated with any obvious phenotypic changes, indicating that GGT1 functions cannot be compensated by those of GGT5 [19][20][21]. The predominance of GGT1 among the GGT family proteins in terms of GSH metabolism is further supported by a recent genetic analysis of two siblings with a GGT deficiency (i.e., glutathionuria, OMIM 231950) [22]. Whole-genome sequencing identified a large homozygous intragenic deletion in GGT1 causing glutathionuria in the patients. Other potentially active enzymes in the GGT family include GGT7, which shares amino acid similarities with GGT1. However, neither its expression pattern nor its functions have been determined [15]. Reports on the protein expression analysis of GGT7 are only limited to ovarian cancer and glioblastoma and have produced conflicting results; in the former, strong GGT7 expression was associated with higher serum GGT, which was in turn associated with an advanced tumor stage and a worse prognosis while, in the latter, strong GGT7 expression was associated with a better prognosis [23][24]. Further studies are needed to clarify the behavior of these proteins, given that GGT7 may have different enzymatic functions as it shares only 47% and 52% of its amino acid sequence with GGT1 and GGT5, respectively, and has high variation in its light chain 15. Taken together, GGT1 is the most extensively studied catalytic enzyme among the GGT family proteins and is ubiquitously expressed in the human body [25].
A recombinant human GGT1 protein was identified as a heterodimer consisting of two glycosylated subunits with a mean molecular mass of 80 kDa and 29 kDa, respectively. The crystal structure of human GGT1 was subsequently obtained by high-resolution X-ray crystallography [26][27]. The active site of human GGT1 was identified as Thr-381, which can be blocked by competitive inhibitors, including acivicin, azaserine, and 6-diazo-5-oxo-norleucine, all of which are glutamate analogs [28].
2.2. Functions of GGT
GGT is an ectoenzyme in the cellular membrane and is known to cleave the γ-glutamyl bond of most γ-glutamyl peptide substrates, including GSH, glutathione S-drug conjugates, and leukotriene C4, resulting in a broad range of protective activities against oxidative stress, drugs, and inflammation [29][30][31]. Among the molecules cleaved by GGT is GSH, a tripeptide consisting of glutamate, cysteine, and glycine that functions as a major intracellular water-soluble antioxidant [2]. Glutamate is linked via a peptide bond through its γ-carboxyl to cysteine. This unusual peptide bond makes GSH resistant to degradation by most peptidases. Among its three constituent amino acids, cysteine is the least available, and thus GGT and the γ-glutamyl cycle play an important role in the metabolism of GSH, especially under cysteine-limited circumstances [32]. In fact, experimentally overexpressed GGT in melanoma cells promoted tumor growth through an intertissue flow of GSH and increased resistance to oxidative stress by recycling cysteine from the extracellular GSH [33][34]. Likewise, transfection of melanoma cells with GGT cDNA resulted in resistance to cisplatin, a major antitumor agent, presumably because of high intracellular cysteinyl–glycine levels resulting from GGT-mediated catabolism of extracellular GSH [35].
The extracellular domain of GGT cleaves the γ-glutamyl bond of GSH and releases cysteinyl–glycine and a γ-glutamyl amino acid at the initial step of the γ-glutamyl cycle (Figure 1). These peptides are subsequently cleaved by γ-glutamylcyclotransferase to yield 5-oxoproline, a free amino acid that is further converted into glutamate by the action of 5-oxoprilinase [3]. This cycle is also important for the resynthesis of intracellular GSH via step-by-step ligation of glutamate, cysteine, and glycine through the sequential action of glutamate–cysteine ligase and glutathione synthetase, leading to the homeostatic maintenance of the intracellular redox potential, as demonstrated in isolated kidney cells [2]. Another key enzyme involved in GSH homeostasis is cation transport regulator 1 (CHAC1), a newly discovered γ-glutamylcyclotransferase involved in the intracellular degradation of GSH [36]. This soluble enzyme is expressed primarily in the cytoplasm and can specifically degrade GSH into 5-oxoproline and cysteinyl-glycine but is inactive with other γ-glutamyl peptides [37]. The shared ability of GGT and CHAC1 to degrade GSH into its component peptides, including cysteine, suggests that they interactively play a scavenging function when cells are exposed to cysteine depletion [36].
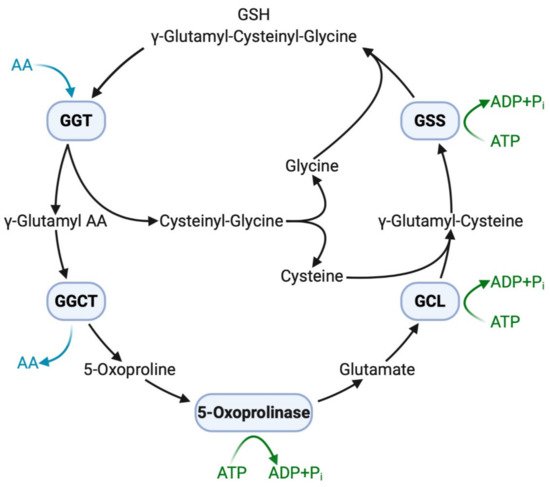
Figure 1. GGT and other enzymes in the γ-glutamyl cycle. Extracellular GSH (γ-glutamyl–cysteinyl–glycine) is broken down on the cell membrane by membrane-bound GGT, making component peptides available to cells with successive reactions with other enzymes. Intracellular GSH is then resynthesized, increasing resistance to oxidative stress. AA, amino acid; ADP, adenosine diphosphate; ATP, adenosine triphosphate; GCL, glutamate-cysteine ligase; GGCT, γ-glutamylcyclotransferase; GGT, γ-glutamyltransferase; GSH, glutathione; GSS, glutathione synthetase; Pi, inorganic phosphate.
The antioxidative properties of GGT have also been attested by evidence showing that GGT transcription is stimulated by a pro-inflammatory cytokine of tumor necrosis factor-α (TNF-α), and that various inhibitors of nuclear factor-κB (NF-κB) prevent overexpression of GGT, confirming that GGT expression induced by TNF-α is mediated by NF-κB [38]. Interestingly, GGT itself can act as a modulator of NF-κB transcriptional activity since it impairs the redox status of thiols that are indispensable for NF-κB DNA binding and gene transactivation [39]. On the other hand, nuclear factor E2-related factor 2 (Nrf2) is involved in an important signaling pathway within the antioxidant response of cancer cells to ROS [40]. Nevertheless, GGT promoter activity is reportedly independent of the Nrf2 signaling pathway, as demonstrated by transfection of cells with small interfering RNA against Nrf2 [41]. Both NF-κB and Nrf2 are induced by ROS and are believed to interfere with one another [42]. GGT located downstream of NF-κB thus promotes proliferation while avoiding apoptosis via Nrf2, eventually leading to carcinogenesis (Figure 2). Apart from its potential role in carcinogenesis, GGT expression is necessary in cell proliferation, migration, and tumor growth because GGT inhibition induces cell cycle arrest with decreased Ki67-positive cells [43]. Thus, GGT appears to be involved in both the early stages of carcinogenesis and the late stages of tumor progression.
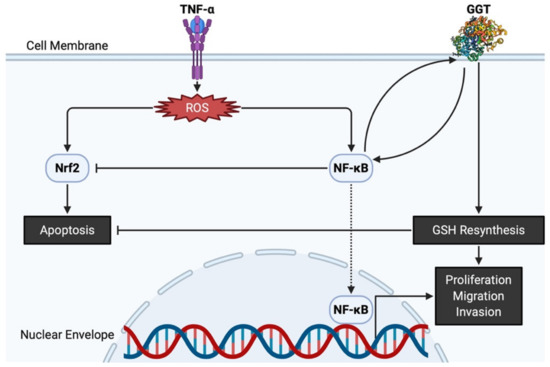
Figure 2. GGT and related molecules in carcinogenesis and tumor progression. TNF-α produces ROS, leading to Nrf2 and NF-κB overpresentation. GGT interacts with NF-κB and helps NF-κB interfere with apoptosis induced by Nrf2. Both nuclear translocation of NF-κB and intracellular resynthesis of GSH, each supported by the enzymatic activities of GGT, have a positive effect on the proliferation, migration, and invasion of cancer cells. GGT, γ-glutamyltransferase; GSH, glutathione; NF-κB, nuclear factor-κB; Nrf2, nuclear factor E2-related factor 2; ROS, reactive oxygen species; TNF-α, tumor necrosis factor-α.
2.3. Expression of GGT in Normal Cells and Cancer Cells in Urogenital Organs
The physiological function of GGT has been described most clearly in the kidney, where GGT localized on the luminal surface of the proximal tubule cells prevents GSH excretion into the urine by initiating the cleavage of GSH into its constituent amino acids, which can then be reabsorbed [25]. Positive immunostaining for GGT was also observed in secretory epithelial cells of the prostatic ducts and acini although the underlying basal epithelial cells were negative for GGT [44]. GGT expression in the urinary tract is very low and only focal expression has been observed in the stroma under the urothelium [25]. Nonetheless, increased expression of GGT was reported in experimentally induced UC in rat urinary bladder treated with the carcinogen N-butyl-N-4-hydroxybutylnitrosamine, suggesting a more direct relationship with carcinogenesis in GGT than in other enzymes involved in drug metabolism [45]. Overexpression of GGT is not a specific marker for UC but is universally observed in tumors arising from different urogenital organs, including the urethra, prostate, and kidney, according to a previous comprehensive immunohistochemical analysis using an affinity-purified polyclonal antibody against peptides corresponding to the C-terminus of the heavy subunit of human GGT [46].
In general, malignant cells maintained the same phenotype as their normal counterparts in terms of GGT expression; most of the cancer types derived from GGT-positive organs were strongly positive for GGT. Indeed, PC arising from the GGT-positive secretory epithelial cells was positive for GGT while benign prostatic hyperplasia showed weak apical expression of GGT [44]. A partial explanation for the overexpression of GGT in PC lies in the fact that androgens can elevate GGT mRNA expression under the regulation of polyomavirus enhancer activator 3, a transcriptional factor, resulting in 500–800-fold greater GGT activity in normal seminal plasma and prostatic fluid than in normal serum [47][48]. In the aforementioned comprehensive immunohistochemical analysis, clear cell RCC arising from GGT-positive proximal renal tubule epithelial cells was strongly GGT-positive with prominent membranous staining patterns [25]. A more recent immunohistochemical analysis using surgically resected RCC specimens demonstrated that 57 out of 65 cases (not including chromophobe RCC) showed moderate to strong GGT expression regardless of their Fuhrman grade [11]. On the other hand, chromophobe RCC, hypothetically arising from GGT-negative distal renal tubule epithelial cells, was completely negative for GGT in contrast to clear cell RCC [25]. In accordance with GGT protein expression, GGT mRNA expression was found to be > 100-fold lower in chromophobe RCC than in normal kidney and clear cell RCC [49]. These findings suggested that GGT expression levels in GU cancer, as in other organs, reflects the phenotype of the original cell types.
3. Potential Clinical Application of GGT Inhibitors
The present section focuses on the possible clinical application of targeted treatment strategies against GGT utilizing several competitive and uncompetitive inhibitors.
3.1. Competitive GGT Inhibitors
Acivicin, a glutamine analogue, inhibits the catalytic activities of GGT [50] and significantly decreases both the rate of intracellular GSH replenishment and the maximum intracellular GSH content [33]. Following the discovery of acivicin, several in vitro and in vivo studies demonstrated acivicin’s therapeutic potential against various types of cancer, including PC, colon cancer, and melanoma [47][51][52]. During the 1980s and 1990s, acivicin was tested for its clinical relevance in phase I and phase II trials in patients with various advanced malignancies [53][54][55][56][57][58][59][60]. Despite the objective antitumor activity observed in some trials, acivicin failed to be approved for clinical use because of the potential for severe toxicity, such as lethal myelosuppression and neurotoxicity, although the causes of these adverse events were not clearly known at the time. However, the detailed mechanism of growth inhibition by acivicin has recently been confirmed based on proteomic profiling of cancer cells. The present study demonstrated that acivicin inhibits aldehyde dehydrogenase 4A1 activity by binding to its catalytic site and does not exert its clinical effects through GGT inhibition, as acivicin has a very low affinity for human GGT [61]. Other glutamine analogues (e.g., the serine–borate complex and γ-phosphono diester analogues of glutamate) and glutamate derivatives or their analogues (e.g., sulfur derivatives of L-glutamic acid and 6-diazo-5-oxo-norleucine) have also been proposed; however, they cause similar toxicity-related problems because they not only inhibit GGT activity but also other essential glutamine metabolizing enzymes, including glutaminases and L-asparagine synthetase [62][63][64][65].
3.2. Uncompetitive GGT Inhibitors
Several novel inhibitors have been developed to overcome the low affinity and high toxicity of existing GGT inhibitors. OU749, a lead compound, is an uncompetitive inhibitor that occupies the acceptor site but not the γ-glutamyl site of GGT [66]. It is worth noting that OU749 is more than 150-fold less toxic than acivicin but has 7–10-fold greater inhibitory potency against GGT isolated from human kidney than GGT isolated from rat or mouse kidney. This ability of OU749 has also been validated under in vivo conditions using GGT-positive cell lines; OU749 blocked GGT catabolism of GSH, providing a basis for further development of a novel therapeutic agent capable of inhibiting GGT via a mechanism distinct from that of the toxic glutamine analogue [67]. Ovothiols, which are histidine-derived thiols isolated from sea urchin eggs, are known to protect the eggs from high oxidative stress at fertilization and may induce autophagy-dependent cell death in human hepatic cancer cells with high GGT expression while leaving normal cells unaffected [68]. Current studies have revealed that the uncompetitive GGT-inhibitory properties of ovothiols produced by marine invertebrates provide great promise for improving molecular-targeted therapy for GGT-positive malignancies, including GU cancer [69][70]. Clinical trials and optimization of these less toxic GGT inhibitors are warranted for the development of novel treatments.
This entry is adapted from the peer-reviewed paper 10.3390/antiox10040549
References
- Meister, A.; Anderson, M.E. Glutathione. Annu Rev. Biochem. 1983, 52, 711–760.
- Hagen, T.M.; Aw, T.Y.; Jones, D.P. Glutathione uptake and protection against oxidative injury in isolated kidney cells. Kidney Int. 1988, 34, 74–81.
- Meister, A. On the enzymology of amino acid transport. Science 1973, 180, 33–39.
- Koenig, G.; Seneff, S. Gamma-glutamyltransferase: A predictive biomarker of cellular antioxidant inadequacy and disease risk. Dis. Markers 2015, 2015, 818570.
- Ou, Y.; Huang, J.; Yang, L. The prognostic significance of pretreatment serum gamma-glutamyltranspeptidase in primary liver cancer: A meta-analysis and systematic review. Biosci. Rep. 2018, 38.
- Sun, P.; Li, Y.; Chang, L.; Tian, X. Prognostic and clinicopathological significance of Gamma-Glutamyltransferase in patients with hepatocellular carcinoma: A PRISMA-compliant meta-analysis. Medicine 2019, 98, e15603.
- Ramankulov, A.; Lein, M.; Kristiansen, G.; Meyer, H.A.; Loening, S.A.; Jung, K. Elevated plasma osteopontin as marker for distant metastases and poor survival in patients with renal cell carcinoma. J. Cancer Res. Clin. Oncol. 2007, 133, 643–652.
- Hofbauer, S.L.; Stangl, K.I.; De Martino, M.; Lucca, I.; Haitel, A.; Shariat, S.F.; Klatte, T. Pretherapeutic gamma-glutamyltransferase is an independent prognostic factor for patients with renal cell carcinoma. Br. J. Cancer 2014, 111, 1526–1531.
- Dalpiaz, O.; Pichler, M.; Mrsic, E.; Reitz, D.; Krieger, D.; Venturino, L.; Bezan, A.; Stojakovic, T.; Pummer, C.; Zigeuner, R.; et al. Preoperative serum-gamma-glutamyltransferase (GGT) does not represent an independent prognostic factor in a European cohort of patients with non-metastatic renal cell carcinoma. J. Clin. Pathol. 2015, 68, 547–551.
- Luo, C.; Xu, B.; Fan, Y.; Yu, W.; Zhang, Q.; Jin, J. Preoperative gamma-glutamyltransferase is associated with cancer-specific survival and recurrence-free survival of nonmetastatic renal cell carcinoma with venous tumor thrombus. Biomed. Res. Int. 2017, 2017, 3142926.
- Takemura, K.; Yuasa, T.; Inamura, K.; Amori, G.; Koga, F.; Board, P.G.; Yonese, J. Impact of serum γ-glutamyltransferase on overall survival in patients with metastatic renal cell carcinoma in the era of targeted therapy. Target. Oncol. 2020, 15, 347–356.
- Ishiyama, Y.; Kondo, T.; Tachibana, H.; Ishihara, H.; Fukuda, H.; Yoshida, K.; Takagi, T.; Iizuka, J.; Tanabe, K. Predictive role of γ-glutamyltransferase in patients receiving nivolumab therapy for metastatic renal cell carcinoma. Int. J. Clin. Oncol. 2021, 26, 552–561.
- Takemura, K.; Ito, M.; Nakanishi, Y.; Kataoka, M.; Sakamoto, K.; Suzuki, H.; Tobisu, K.-I.; Koga, F. Serum γ-glutamyltransferase as a prognostic biomarker in metastatic castration-resistant prostate cancer treated with enzalutamide. Anticancer Res. 2019, 39, 5773–5780.
- Takemura, K.; Fukushima, H.; Ito, M.; Kataoka, M.; Nakanishi, Y.; Sakamoto, K.; Suzuki, H.; Tobisu, K.-I.; Koga, F. Prognostic significance of serum γ-glutamyltransferase in patients with advanced urothelial carcinoma. Urol. Oncol. 2019, 37, 108–115.
- Heisterkamp, N.; Groffen, J.; Warburton, D.; Sneddon, T.P. The human gamma-glutamyltransferase gene family. Hum. Genet. 2008, 123, 321–332.
- Heisterkamp, N.; Rajpert-De Meyts, E.; Uribe, L.; Forman, H.J.; Groffen, J. Identification of a human gamma-glutamyl cleaving enzyme related to, but distinct from, gamma-glutamyl transpeptidase. Proc. Natl. Acad. Sci. USA 1991, 88, 6303–6307.
- Hanigan, M.H.; Gillies, E.M.; Wickham, S.; Wakeham, N.; Wirsig-Wiechmann, C.R. Immunolabeling of gamma-glutamyl transferase 5 in normal human tissues reveals that expression and localization differ from gamma-glutamyl transferase 1. Histochem. Cell Biol. 2015, 143, 505–515.
- Wickham, S.; West, M.B.; Cook, P.F.; Hanigan, M.H. Gamma-glutamyl compounds: Substrate specificity of gamma-glutamyl transpeptidase enzymes. Anal. Biochem. 2011, 414, 208–214.
- Harding, C.O.; Williams, P.; Wagner, E.; Chang, D.S.; Wild, K.; Colwell, R.E.; Wolff, J.A. Mice with genetic gamma-glutamyl transpeptidase deficiency exhibit glutathionuria, severe growth failure, reduced life spans, and infertility. J. Biol. Chem. 1997, 272, 12560–12567.
- Levasseur, R.; Barrios, R.; Elefteriou, F.; Glass, D.A.; Lieberman, M.W.; Karsenty, G. Reversible skeletal abnormalities in gamma-glutamyl transpeptidase-deficient mice. Endocrinology 2003, 144, 2761–2764.
- Shi, Z.Z.; Han, B.; Habib, G.M.; Matzuk, M.M.; Lieberman, M.W. Disruption of gamma-glutamyl leukotrienase results in disruption of leukotriene D(4) synthesis in vivo and attenuation of the acute inflammatory response. Mol. Cell Biol. 2001, 21, 5389–5395.
- Darin, N.; Leckstrom, K.; Sikora, P.; Lindgren, J.; Almen, G.; Asin-Cayuela, J. Gamma-glutamyl transpeptidase deficiency caused by a large homozygous intragenic deletion in GGT1. Eur. J. Hum. Genet. 2018, 26, 808–817.
- Grimm, C.; Hofstetter, G.; Aust, S.; Mutz-Dehbalaie, I.; Bruch, M.; Heinze, G.; Rahhal-Schupp, J.; Reinthaller, A.; Concin, N.; Polterauer, S. Association of gamma-glutamyltransferase with severity of disease at diagnosis and prognosis of ovarian cancer. Br. J. Cancer 2013, 109, 610–614.
- Bui, T.T.; Nitta, R.T.; Kahn, S.A.; Razavi, S.M.; Agarwal, M.; Aujla, P.; Gholamin, S.; Recht, L.; Li, G. γ-Glutamyl transferase 7 is a novel regulator of glioblastoma growth. BMC Cancer 2015, 15, 225.
- Hanigan, M.H.; Frierson, H.F. Immunohistochemical detection of gamma-glutamyl transpeptidase in normal human tissue. J. Histochem. Cytochem. 1996, 44, 1101–1108.
- Oster, T.; Visvikis, A.; Thioudellet, C.; Fournel-Gigleux, S.; Wellman, M.; Siest, G. Establishment of a V79 transfected cell line highly producing recombinant human gamma-glutamyltransferase. Toxicology 1993, 82, 151–167.
- West, M.B.; Chen, Y.; Wickham, S.; Heroux, A.; Cahill, K.; Hanigan, M.H.; Mooers, B.H. Novel insights into eukaryotic γ-glutamyltranspeptidase 1 from the crystal structure of the glutamate-bound human enzyme. J. Biol. Chem. 2013, 288, 31902–31913.
- Terzyan, S.S.; Burgett, A.W.; Heroux, A.; Smith, C.A.; Mooers, B.H.; Hanigan, M.H. Human γ-glutamyl transpeptidase 1: Structures of the free enzyme, inhibitor-bound tetrahedral transition states, and glutamate-bound enzyme reveal novel movement within the active site during catalysis. J. Biol. Chem. 2015, 290, 17576–17586.
- Meister, A.; Tate, S.S.; Griffith, O.W. Gamma-glutamyl transpeptidase. Methods Enzymol. 1981, 77, 237–253.
- Lewis, R.A.; Austen, K.F.; Soberman, R.J. Leukotrienes and other products of the 5-lipoxygenase pathway. Biochemistry and relation to pathobiology in human diseases. N. Engl. J. Med. 1990, 323, 645–655.
- Angeli, V.; Tacito, A.; Paolicchi, A.; Barsacchi, R.; Franzini, M.; Baldassini, R.; Vecolid, C.; Pompella, A.; Bramanti, E.; Bramanti, E. A kinetic study of gamma-glutamyltransferase (GGT)-mediated S-nitrosoglutathione catabolism. Arch. Biochem. Biophys. 2009, 481, 191–196.
- Meister, A. The Liver: Biology and Pathobiology, 2nd ed.; Raven Press: New York, NY, USA, 1988.
- Obrador, E.; Carretero, J.; Ortega, A.; Medina, I.; Rodilla, V.; Pellicer, J.A.; Estrela, J.M. Gamma-Glutamyl transpeptidase overexpression increases metastatic growth of B16 melanoma cells in the mouse liver. Hepatology 2002, 35, 74–81.
- Giommarelli, C.; Corti, A.; Supino, R.; Favini, E.; Paolicchi, A.; Pompella, A.; Zunino, F. Cellular response to oxidative stress and ascorbic acid in melanoma cells overexpressing gamma-glutamyltransferase. Eur. J. Cancer 2008, 44, 750–759.
- Franzini, M.; Corti, A.; Lorenzini, E.; Paolicchi, A.; Pompella, A.; De Cesare, M.; Perego, P.; Gatti, L.; Leone, R.; Apostoli, P.; et al. Modulation of cell growth and cisplatin sensitivity by membrane gamma-glutamyltransferase in melanoma cells. Eur. J. Cancer 2006, 42, 2623–2630.
- Mungrue, I.N.; Pagnon, J.; Kohannim, O.; Gargalovic, P.S.; Lusis, A.J. CHAC1/MGC4504 is a novel proapoptotic component of the unfolded protein response, downstream of the ATF4-ATF3-CHOP cascade. J. Immunol. 2009, 182, 466–476.
- Kumar, A.; Tikoo, S.; Maity, S.; Sengupta, S.; Kaur, A.; Bachhawat, A.K. Mammalian proapoptotic factor ChaC1 and its homologues function as γ-glutamyl cyclotransferases acting specifically on glutathione. EMBO Rep. 2012, 13, 1095–1101.
- Reuter, S.; Schnekenburger, M.; Cristofanon, S.; Buck, I.; Teiten, M.H.; Daubeuf, S.; Serge, E.; Dicato, M.; Aggarwal, B.B.; Visvikis, A.; et al. Tumor necrosis factor alpha induces gamma-glutamyltransferase expression via nuclear factor-kappaB in cooperation with Sp1. Biochem. Pharmacol. 2009, 77, 397–411.
- Dominici, S.; Visvikis, A.; Pieri, L.; Paolicchi, A.; Valentini, M.A.; Comporti, M.; Pompella, A. Redox modulation of NF-kappaB nuclear translocation and DNA binding in metastatic melanoma. The role of endogenous and gamma-glutamyl transferase-dependent oxidative stress. Tumori 2003, 89, 426–433.
- Kabel, A.M.; Atef, A.; Estfanous, R.S. Ameliorative potential of sitagliptin and/or resveratrol on experimentally-induced clear cell renal cell carcinoma. Biomed. Pharmacother. 2018, 97, 667–674.
- Huseby, N.E.; Ravuri, C.; Moens, U. The proteasome inhibitor lactacystin enhances GSH synthesis capacity by increased expression of antioxidant components in an Nrf2-independent, but p38 MAPK-dependent manner in rat colorectal carcinoma cells. Free Radic. Res. 2016, 50, 1–13.
- Li, W.; Khor, T.O.; Xu, C.; Shen, G.; Jeong, W.S.; Yu, S.; Kong, A.N. Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem. Pharmacol. 2008, 76, 1485–1489.
- Bansal, A.; Sanchez, D.J.; Nimgaonkar, V.; Sanchez, D.; Riscal, R.; Skuli, N.; Simon, M.C. Gamma-glutamyltransferase 1 promotes clear cell renal cell carcinoma initiation and progression. Mol. Cancer Res. 2019, 17, 1881–1892.
- Frierson, H.F.; Theodorescu, D.; Mills, S.E.; Hanigan, M.H. Gamma-Glutamyl transpeptidase in normal and neoplastic prostate glands. Mod. Pathol. 1997, 10, 1–6.
- Tsuda, H.; Inoue, T.; Asamoto, M.; Fukushima, S.; Ito, N.; Okamura, T.; Ohtaguro, K.; Washida, H.; Satoh, K.; Amelizad, Z.; et al. Comparison of enzyme phenotypes in human bladder tumours and experimentally induced hyperplastic and neoplastic lesions of the rat urinary bladder. A combined histochemical and immunohistochemical approach. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1989, 56, 307–316.
- Hanigan, M.H.; Frierson, H.F.; Swanson, P.E.; De Young, B.R. Altered expression of gamma-glutamyl transpeptidase in human tumors. Hum. Pathol. 1999, 30, 300–305.
- Ripple, M.O.; Pickhardt, P.A.; Wilding, G. Alteration in gamma-glutamyl transpeptidase activity and messenger RNA of human prostate carcinoma cells by androgen. Cancer Res. 1997, 57, 2428–2433.
- Lan, Z.J.; Palladino, M.A.; Rudolph, D.B.; Labus, J.C.; Hinton, B.T. Identification, expression, and regulation of the transcriptional factor polyomavirus enhancer activator 3, and its putative role in regulating the expression of gamma-glutamyl transpeptidase mRNA-IV in the rat epididymis. Biol. Reprod. 1997, 57, 186–193.
- Priolo, C.; Khabibullin, D.; Reznik, E.; Filippakis, H.; Ogórek, B.; Kavanagh, T.R.; Nijmeh, J.; Herbert, Z.T.; Asara, J.M.; Kwiatkowski, D.J.; et al. Impairment of gamma-glutamyl transferase 1 activity in the metabolic pathogenesis of chromophobe renal cell carcinoma. Proc. Natl. Acad. Sci. USA 2018, 115, E6274–E6282.
- Tate, S.S.; Meister, A. Affinity labeling of gamma-glutamyl transpeptidase and location of the gamma-glutamyl binding site on the light subunit. Proc. Natl. Acad. Sci. USA 1977, 74, 931–935.
- Fischer, P.H.; Pamukcu, R.; Bittner, G.; Willson, J.K. Enhancement of the sensitivity of human colon cancer cells to growth inhibition by acivicin achieved through inhibition of nucleic acid precursor salvage by dipyridamole. Cancer Res. 1984, 44, 3355–3359.
- Mena, S.; Benlloch, M.; Ortega, A.; Carretero, J.; Obrador, E.; Asensi, M.; Petschen, I.; Brown, B.D.; Estrela, J.M. Bcl-2 and glutathione depletion sensitizes B16 melanoma to combination therapy and eliminates metastatic disease. Clin. Cancer Res. 2007, 13, 2658–2666.
- McGovren, J.P.; Pratt, E.A.; Belt, R.J.; Taylor, S.A.; Benjamin, R.S.; Ardalan, B.; Ohnuma, T. Pharmacokinetic and biochemical studies on acivicin in phase I clinical trials. Cancer Res. 1985, 45, 4460–4463.
- Willson, J.K.; Fischer, P.H.; Tutsch, K.; Alberti, D.; Simon, K.; Hamilton, R.D.; Bruggink, J.; Koeller, J.M.; Tormey, D.C.; Earhart, R.H.; et al. Phase I clinical trial of a combination of dipyridamole and acivicin based upon inhibition of nucleoside salvage. Cancer Res. 1988, 48, 5585–5590.
- Earhart, R.H.; Khandekar, J.D.; Faraggi, D.; Schinella, R.A.; Davis, T.E. Phase II trial of continuous drug infusions in advanced ovarian carcinoma: Acivicin versus vinblastine. Investig. New Drugs. 1989, 7, 255–260.
- Falkson, G.; Cnaan, A.; Simson, I.W.; Dayal, Y.; Falkson, H.; Smith, T.J.; Haller, D.G. A randomized phase II study of acivicin and 4’deoxydoxorubicin in patients with hepatocellular carcinoma in an Eastern Cooperative Oncology Group study. Am. J. Clin. Oncol. 1990, 13, 510–515.
- Taylor, S.A.; Crowley, J.; Pollock, T.W.; Eyre, H.J.; Jaeckle, C.; Hynes, H.E.; Stephens, R.L. Objective antitumor activity of acivicin in patients with recurrent CNS malignancies: A Southwest Oncology Group trial. J. Clin. Oncol. 1991, 9, 1476–1479.
- Bonomi, P.; Finkelstein, D.; Chang, A. Phase II trial of acivicin versus etoposide-cisplatin in non-small cell lung cancer. An Eastern Cooperative Oncology Group study. Am. J. Clin. Oncol. 1994, 17, 215–217.
- Baruchel, S.; Bernstein, M.; Whitehead, V.M.; Devine, S.; Bell, B.; Dubowy, R.; Grier, H.; Kretschmar, C.; Langevin, A.-M.; Vietti, T. A phase I study of acivicin in refractory pediatric solid tumors. A Pediatric Oncology Group study. Investig. New Drugs. 1995, 13, 211–216.
- Hidalgo, M.; Rodriguez, G.; Kuhn, J.G.; Brown, T.; Weiss, G.; MacGovren, J.P.; Von Hoff, D.D.; Rowinsky, E.K. A Phase I and pharmacological study of the glutamine antagonist acivicin with the amino acid solution aminosyn in patients with advanced solid malignancies. Clin. Cancer Res. 1998, 4, 2763–2770.
- Kreuzer, J.; Bach, N.C.; Forler, D.; Sieber, S.A. Target discovery of acivicin in cancer cells elucidates its mechanism of growth inhibition†Electronic supplementary information (ESI) available: Synthesis, cloning, protein expression, purification and biochemical assays. Chem. Sci. 2014, 6, 237–245.
- Tate, S.S.; Meister, A. Serine-borate complex as a transition-state inhibitor of gamma-glutamyl transpeptidase. Proc. Natl. Acad. Sci. USA 1978, 75, 4806–4809.
- Han, L.; Hiratake, J.; Kamiyama, A.; Sakata, K. Design, synthesis, and evaluation of gamma-phosphono diester analogues of glutamate as highly potent inhibitors and active site probes of gamma-glutamyl transpeptidase. Biochemistry 2007, 46, 1432–1447.
- Lherbet, C.; Keillor, J.W. Probing the stereochemistry of the active site of gamma-glutamyl transpeptidase using sulfur derivatives of l-glutamic acid. Org. Biomol. Chem. 2004, 2, 238–245.
- Terzyan, S.S.; Cook, P.F.; Heroux, A.; Hanigan, M.H. Structure of 6-diazo-5-oxo-norleucine-bound human gamma-glutamyl transpeptidase 1, a novel mechanism of inactivation. Protein Sci. 2017, 26, 1196–1205.
- King, J.B.; West, M.B.; Cook, P.F.; Hanigan, M.H. A novel, species-specific class of uncompetitive inhibitors of gamma-glutamyl transpeptidase. J. Biol. Chem. 2009, 284, 9059–9065.
- Wickham, S.; Regan, N.; West, M.B.; Thai, J.; Cook, P.F.; Terzyan, S.S.; Kai Li, P.; Hanigan, M.H. Inhibition of human γ-glutamyl transpeptidase: Development of more potent, physiologically relevant, uncompetitive inhibitors. Biochem. J. 2013, 450, 547–557.
- Russo, G.L.; Russo, M.; Castellano, I.; Napolitano, A.; Palumbo, A. Ovothiol isolated from sea urchin oocytes induces autophagy in the Hep-G2 cell line. Mar. Drugs 2014, 12, 4069–4085.
- Milito, A.; Brancaccio, M.; Lisurek, M.; Masullo, M.; Palumbo, A.; Castellano, I. Probing the interactions of sulfur-containing histidine compounds with human gamma-glutamyl transpeptidase. Mar. Drugs 2019, 17, 650.
- Brancaccio, M.; Russo, M.; Masullo, M.; Palumbo, A.; Russo, G.L.; Castellano, I. Sulfur-containing histidine compounds inhibit γ-glutamyl transpeptidase activity in human cancer cells. J. Biol. Chem. 2019, 294, 14603–14614.