The role of three members of the Chaperone Systems (CS)—heat shock protein (Hsp)60, Hsp70, and Hsp90—in Immune Systems (IS) modulation and neuroinflammation. These three chaperones occur intra- and extracellularly, with the latter being the most likely involved in neuroinflammation because they can interact with the IS.
- chaperone system
- molecular chaperones
- Neurodegeneration
- Inflammation
- Alzheimer’s Disease
- Parkinson’s Disease
- myotrophic Lateral Sclerosis
- Hsp60
- Hsp70
- Hsp90
1. Immunomodulatory Function of Extracellular Hsp60, Hsp70, and Hsp90
Hsp60, Hsp70, and Hsp90 interact with the immune system in many ways and thereby have an impact on neurodegenerative diseases. Extracellular Hsp60, Hsp70, and Hsp90 influence both the innate and the adaptive immune responses. Generally, extracellular Hsp–receptor interaction involves specific receptors expressed on macrophages and dendritic and microglia cells, including toll-like receptors (TLRs), scavenger receptors (SR), and other molecules [1]. For example, Hsp70 and Hsp90 can interact with the SR LOX-1 [2], and Hsp70 interacts also with multiple members of the SR family [3]. The SR are expressed on different types of cells and they are involved in the binding and internalization of stress proteins [3]. Extracellular Hsp60, Hsp70, and Hsp90 can modulate the innate immune response, causing the secretion of proinflammatory cytokines by APCs [4]. This interaction elicits a proinflammatory response that involves mainly nuclear factor-kappa B (NF-kB). These chaperones are endogenous ligands for TLRs, and by interacting also with CD14 molecules, they can induce the production of cytokines (e.g., interleukin 1 beta (IL-1β), IL-6, inducible isoform of nitric oxide synthase (iNOS)) [5][6]. TLR4 is a receptor expressed on the microglia plasma cell membrane with a key role in the generation of immune responses in the nervous system, responses that are implicated in the development of neurodegenerative disorders [7]. For instance, Hsp60 can mediate neuroinflammation through a MyD88-dependent pathway by interacting with TLR4 on the microglia surface [6] and by inducing the production of proinflammatory factors via microglial LOX-1 [8]. Intrathecal injection of Hsp60 lead to neurodegeneration and demyelination by the activation of TLR4-MyD88 signaling in microglial cells [9]. Hsp70 can interact with microglia, dendritic cells, and macrophages through TLR2 and TLR4, leading to proinflammatory NF-kB activation and its associated pathways [10]. Hsp90 interacts with an extensive list of key mediators involved in pathways regulating inflammatory and immune responses. For example, among the protein clients of Hsp90, there is the receptor-interacting protein (RIP) kinase, which is involved in the innate immune response and in the cell-death signaling pathway. [11] RIP, following TLR4 activation, induces the expression of proinflammatory cytokines by NF-kB signaling [12] (Figure 1).
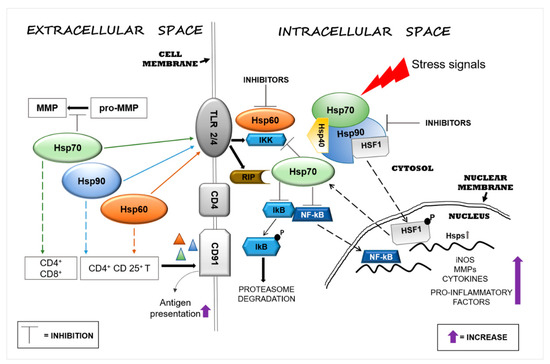
Figure 1. Heat shock protein (Hsp)60, Hsp70, and Hsp90 modulate inflammatory reactions by interacting with factors involved in the regulation of innate and adaptive immune responses. Stressors can activate the immune system and, in turn, promote neurodegeneration by inducing Hsps in brain tissue as a mechanism of protection. Extracellular Hsp60, Hsp70, and Hsp90 interact with receptors present on the surface of cells of the neural tissue’s immune compartment (e.g., microglia) and elicit pro- or anti-inflammatory responses, depending on the local cellular status. The interaction of extracellular Hsp60, Hsp70, and Hsp90 with toll-like receptor (TLR)2/4 induces the activation of the nuclear factor-kappa B (NF-kB) inhibitor protein, which in turn triggers the activation of the NF-kB pathway, promoting an inflammatory response. Hsp60, Hsp70, and Hsp90 form complexes with antigens (represented by triangles) mediating their presentation via the CD91 cell surface receptor on antigen-presenting cells (APCs) [13]. Hsp90 plays a proinflammatory role through the interaction with its client proteins, such as members of the receptor-interacting protein (RIP) kinases and, thereby, activates the NF-kB pathway. Under physiological conditions, intracellular Hsp90, by blocking heat shock factor (HSF)1, prevents the transcription of Hsp genes, such as Hsp70, or other genes that code for anti-inflammatory molecules. The pharmacological inhibition of Hsp90 can lead to upregulation of the transcription of intracellular Hsp70 and of anti-inflammatory molecules by its release. Abbreviations: MMP, matrix metalloproteinase; TLR, toll-like receptor; IKK, inhibitor of κB kinase; RIP, receptor interaction protein; CD, cluster of differentiation; lkB, inhibitory subunit I kappa B-alpha; NF-kB, nuclear factor kappa-light-chain-enhancer of activated B cells; HSF1, heat shock factor 1; P, phosphate; Hsp, heat shock protein; iNOS, inducible isoform of nitric oxide synthase.
Extracellular Hsp60, Hsp70, and Hsp90 can also help antigen presentation in the adaptive immune responses by upregulating the expression of major histocompatibility complex (MHC) molecules and their load [14][15]. Extracellular Hsp70 and Hsp90 complexed with antigens elicit the responses of cluster of differentiation (CD)8+ or CD4+ T cells by adaptive receptors [1], while Hsp60 by itself can stimulate regulatory CD4+ and CD 25+ T cells (Tregs), leading to an immunosuppressive adaptive response without APC participation [1][16]. In addition, the chaperone–peptide complexes can also recognize the CD91 receptor of macrophages/dendritic cells and facilitate antigen presentation [13] (Figure 1). The activation of the adaptive response via Hsp70 might represent a negative reaction for the cell, but it could be considered advantageous for the development of immunological memory in preparation for rapid reaction against subsequent insults [17]. In contrast to the proinflammatory function of extracellular Hsp70, intracellular Hsp70 has an anti-inflammatory effect in the brain, especially when overexpressed following brain damage. Thus, Hsp70 can be anti-inflammatory because it can block the expression of proinflammatory molecules, such as matrix metalloproteinases [18], and it can also promote the reduction or the inhibition of NF-kB activity [19][20] (Figure 1). In addition, intracellular Hsp70 interferes also with genes involved in various neuronal pathways such as transmission of nerve impulses [21]. Therefore, extracellular Hsp70 could in principle have anti-inflammatory and neuroprotective effects similar to those of the intracellular counterpart [22]. Consequently, it is likely that an increase of intracellular Hsp70 will lead to an increase of functional extracellular Hsp70, contributing to the reduction of the inflammation associated with neurodegeneration. Pharmacological increase of the Hsp70 level in neurons and microglia by 17-N-allylamino-17-demethoxygeldanamycin (17-AAG) reduced the hemorrhagic volume in a mouse model of traumatic brain injury [23]. Likewise, 17-AAG inhibition of Hsp90 induced the expression of Hsp70 and Hsp60 [24]. Thus, it may be said that Hsp70, Hsp60, and Hsp90 promote inflammatory responses and, consequently, neuronal damage and are implicated in neuroinflammation and neurotoxicity. Cytosolic Hsp60 has been shown to directly interact with the inhibitor of κB kinase (IKK), promoting activation of NF-kB-dependent gene transcription by tumor necrosis factor-α (TNFα) [25] (Figure 1). Hsp90 can induce a proinflammatory response in different ways, for example, by sequestering the regulator transcriptional factor heat shock factor (HSF)1 and thereby inhibiting the expression of Hsps (e.g., Hsp70) or activating the NF-kB pathway through the activation of its protein clients RIP [26] (Figure 1). In view of these results, Hsp60 and Hsp90 modulators appear as potentially useful agents for controlling inflammation in the nervous system [27][28]. Currently, numerous compounds have been designed to inhibit Hsp90 activity, but few have been developed for Hsp60 [29]. Hsp90 inhibitors have been developed to directly act on the chaperone or on its client proteins. Some inhibitors block the Hsp90 folding activity linked to adenosine triphosphate (ATP)-dependent conformation changes [30], while others inactivate its client proteins via proteasomal degradation [31]. For example, geldanamycin induces the degradation of Hsp90 client proteins of the RIP family with the consequent inhibition of TNF-mediated IkB kinase and NF-kB activation [26]. Furthermore, Hsp90 forms a complex with HSF1, blocking its translocation to the nucleus and, thereby, impedes the upregulation of Hsp70 and other anti-inflammatory molecules [32] (Figure 1). Most of the compounds that inhibit Hsp70 function by targeting its ATP hydrolysis activity or specific cysteine residues [33].
2. Extracellular Hsp60, Hsp70, and Hsp90 in Acute Nervous System Injury and Chronic Neurodegenerative Diseases
Neurodegenerative diseases are accompanied by inflammatory responses aimed at eliminating dead and damaged neuronal cells to restore the compromised area to its normal status [34]. It should be borne in mind that while short-lived inflammatory responses generally have a beneficial effect, excessive and persistent release of inflammatory mediators can be harmful to brain tissue [35]. Moreover, prolonged activation of microglia and astrocytes could also lead to the alteration of their beneficial functions, which they display under normal conditions [36]. Therefore, it is not surprising that neuroinflammation contributes to CNS diseases [37]. Although different in their origins, many neurodegenerative conditions are characterized by shared cellular responses that promote the upregulation of molecular chaperones as the first line of defense against misfolded, dysfunctional, and aggregation-prone proteins [38]. There is increasing evidence for the release of Hsp60, Hsp70, and Hsp90 into the extracellular environment, with functions that are complementary or independent of those of their intracellular counterparts. Since these chaperones lack a secretion signal in their sequences, the mechanisms by which they are released are poorly understood. In vitro and in vivo studies with Hsp60 have unveiled secretion pathways, involving lipid rafts and exosomes, which would explain the presence of Hsp60 in extramitochondrial sites such as interstitial space, cellular membrane, and biological fluids [39]. Similarly, nontraditional secretion mechanisms participate in the membrane delivery and release of Hsp70, involving lipid rafts [40] and lysosomes [41], in line with its role as a lysosomal stabilizer [42]. Secretion of Hsp90 via exosomes depends on its ATPase function and on the open or closed conformational state of the Hsp90 dimer: the open state promotes Hsp90 release via exosomes, whereas the closed state blocks this process [43]. Different types of CNS cells, including neurons and glial cells, can release exosomes with their cargo of specific molecules that could affect the function of acceptor cells [44]. At the extracellular level, Hsp60 is known to contribute to neuroinflammation with possible negative implications: this chaperone is highly expressed in activated microglia, and when released extracellularly, it induces neuroinflammation with neuronal cell death [45]. For this reason, inhibition of Hsp60 expression and its release represents a possible therapeutic mechanism applicable to neurodegenerative diseases. The pro- and anti-inflammatory effects of extracellular Hsp60, Hsp70, and Hsp90 in AD, PD, ALS, HD, and MS are summarized in Table 1 and discussed in the following paragraphs.
Table 1. Anti- and proinflammatory effects of extracellular Hsp60, Hsp70, and Hsp90 in neurodegenerative diseases.
| Disease | Hsp60 | Hsp70 | Hsp90 |
|---|---|---|---|
| Alzheimer’s disease (AD) | Anti- | Anti- | Pro- |
| Parkinson’s disease (PD) | Pro- | Anti- | Pro- |
| Huntington’s disease (HD) | Anti- | Anti- | Pro- |
| Amyotrophic lateral sclerosis (ALS) | Not reported | Anti- | Not reported |
| Multiple sclerosis (MS) | Pro- | Pro- | Pro- |
3.1. Alzheimer’s Disease
AD is a neurodegenerative disorder in which the amyloid-β peptide (Aβ) accumulates in extracellular deposits named plaque, whereas neurofibrillary tangles (NFTs) occur intracellularly with hyperphosphorylated tau [46][47]. Under pro-aggregating conditions (37 °C and stirring), extracellular Hsp60 inhibits the onset of Aβ cross-β-structure formation that typically accompanies the peptide assembly toward higher ordered structures [48]. The hypotheses formulated on the possible role of Hsp60 in the formation of protein deposits are mainly based on its holding activity. For instance, Hsp60 could act as a noncatalytic inhibitor of polypeptide aggregation by sequestering unfolded monomers via hydrophobic interactions. However, the stoichiometric ratio of the Aβpeptide/Hsp60 and the limitation of the methods applied for these measurements put a question mark on the validity of the results. In fact, the inhibition of amyloid formation appears discontinuous when passing from a 75:1 to 50:1 molar ratio. Furthermore, the method used, size-exclusion chromatography, cannot distinguish between Aβ monomers or peptide oligomers of very low molecular weight, such as dimers or trimers, nor can it discriminate between on-pathway and off-pathway species. These data suggest that Hsp60 exerts its inhibitory action only under stress conditions and, in particular, in the presence of other factors such as high temperature and stirring, which favor the formation of on-pathway seeding species. [48]. Higher levels of Hsp60 were found in lymphocytes isolated from AD subjects [49][50]. αβ immunization with peptides derived from Hsp60 induced a decrease of cerebral amyloid burden in a mouse model [51]. Like Hsp60, extracellular Hsp70 also interacts with Aβ oligomers, blocking their oligomerization into fibers and reducing their toxicity [52]. The engineered form of secreted Hsp70 (secHsp70) in Drosophila protects against the toxicity induced by Aβ42 deposits in the extracellular milieu [53]. Exogenous Hsp90 was found to induce microglial activation and to facilitate phagocytosis and clearance of Aβ directly via the TLR4 pathway, but when bound to the Aβ oligomers, it induced the production of IL-6 and TNF-α [54]. In another work, it was revealed that Hsp90 modulates the formation of the STIP1 (or Hsp70/Hsp90 organizing protein (HOP))/PrPC complex, which inhibits the neuroprotective role of STIP1 against amyloid-beta peptide [55]. However, it is still unclear whether extracellular Hsp70/Hsp90/STIP1 in AD brain exists separately or as a complex with the Aβ aggregate [55]. All these observations indicate that the understanding of Hsp90′s role in neurodegeneration deserves further investigation.
3.2. Parkinson’s Disease
PD is characterized by movement disorders and loss of dopaminergic neurons in the brain’s substantia nigra pars compacta [56][57]. The disease is also characterized by aggregated α-synuclein that forms nuclear inclusions called Lewy bodies [58]. A study in yeast cells has shown that null mutations in the Hsp60 gene are linked with defects in the folding of mitochondrial proteins, with accumulations of misfolded peptides analogous to the α-synuclein aggregates of PD [59]. Hsp60, Hsp70, and Hsp90 interact with α-synuclein in the Lewy bodies in PD patients. These inclusions consist not only of α-synuclein aggregates but also contain molecular chaperones which have been sequestered in the aggregates while attempting to impede or correct protein misfolding and aggregation [60][61]. This sequestration leads to a deficit of chaperones available for maintaining protein homeostasis, namely, a chaperonopathy by defect occurs, which contributes to the aggravation of the pathologic process leading to neurodegeneration. The interaction between Hsp70 and α-synuclein involves the central hydrophobic region of the pathological protein and the substrate-binding domain Hsp70 and is crucial for inhibiting assembly before the elongation stage [62]. The neuroprotective function of overexpressed Hsp70 has been confirmed in experimental models in vivo [63]. There is less information regarding the protective role of Hsp90 in the regulation of α-synuclein aggregation. Like in AD, Hsp60, Hsp70, and Hsp90 contribute to neuronal toxicity in PD. Hsp90 abolishes the binding of α-synuclein to vesicles and promotes the formation of fibrils [64]. In in vivo and in vitro models of PD, it was found that Hsp60 expression gradually decreased after 6-hydroxydopamine (6-OHDA) injection into dopaminergic neurons (DA). This result may be explained by the release of Hsp60 by the damaged neurons, as suggested by its presence in the cell culture medium [65]. In PD models and patients, activation of microglia plays a key role in the release of proinflammatory factors that aggravate the loss of DA neurons [66]. Astrocytes, which are the predominant glial cell type in the CNS, are also critically affected by stressors. The expression of Hsp60 on the surface of activated microglia suggests that Hsp60 is involved in the progression of PD. Extracellular release of Hsp60 from CNS cells undergoing necrotic or apoptotic death activates microglia in a TLR4- and MyD88-dependent manner [6]. Hsp60 was released from degenerated neurons to activate microglia in a rat PD model, providing a novel idea for developing a therapeutic strategy to slow or stop PD progression by preventing the release of Hsp60 or interfering with the interaction between Hsp60 and microglia [65].
3.3. Amyotrophic Lateral Sclerosis
ALS is a chronic inflammatory demyelinating disease that affects motor neurons and is characterized by atrophy and paralysis of muscles, with progressive aggravation over the years [67]. This disease occurs sporadically, but a small percentage is familial with mutations in specific genes, such as the gene encoding the free-radical-scavenging enzyme superoxide dismutase-1 (SOD1) [68]. An important aspect of SOD1-associated ALS is the deposition of SOD1 in large insoluble aggregates in motor neurons. The SOD1 mutated protein mediates the induction of the disease through the dysregulation of the heat shock response (HSR)–apoptosis axis [69]. The development of ALS is linked to the formation of intracellular aggregates of misfolded proteins [67]. Few data are available regarding the involvement of molecular chaperones in ALS onset. Motor neurons of ALS patients have an intrinsic deficit in the ability to activate the HSR and, consequently, do not readily regulate Hsp expression, as shown, for example, for Hsp70 [70]. It has been observed that the Hsp70/Hsp40 pair is complexed with the mutant form of the SOD1 protein in cultured neuronal cells [71]. However, data indicate that the increase of Hsp70 level alone is not sufficient to ameliorate mutant SOD1-protein-mediated toxicity in mouse models [72]. Histamine is neuroprotective through the HSR in motor neurons and microglia cell cultures, and in vivo in spinal cord and cortex from symptomatic SOD1-G93A mice [73]. These results emphasize the relevance of histidine-induced Hsp70 stimulation for preserving motor function [73]. Further, the intraperitoneal administration of human recombinant exogenous Hsp70 increased lifespan, delayed the onset of symptoms, preserved locomotor function, and prolonged motoneuron survival in a mouse model of ALS [74]. Extracellular Hsp70 stimulates the survival of neurons following injury [75] and overexpressed Hsp70 induces the survival of astrocytes [76]. Under stress, astrocytes increase the release of exosomes enriched in Hsp70, with positive implications on the survival of nearby neurons [77]. Interestingly, exosomes derived from cancer cells express Hsp70 on their surface, which allows their interaction with target cells carrying surface Hsp receptors [29]. Hsp70 (DnaJC5/Hsc70 complex) is also believed to be involved in the extracellular release of proteins associated with neurodegenerative disease as part of its chaperoning functions [78]. Exogenous Hsp70 protects from oxidative damage death in motor neurons through binding and sequestration of toxic proteins [79].
This entry is adapted from the peer-reviewed paper 10.3390/app11020736
References
- Calderwood, S.K.; Gong, J.; Murshid, A. Extracellular HSPs: The Complicated Roles of Extracellular HSPs in Immunity. Front. Immunol. 2016, 7, 159.
- Murshid, A.; Theriault, J.; Gong, J.; Calderwood, S.K. Investigating receptors for extracellular heat shock proteins. Methods Mol. Biol. 2011, 787, 289–302.
- Thériault, J.R.; Adachi, H.; Calderwood, S.K. Role of scavenger receptors in the binding and internalization of heat shock protein 70. J. Immunol. 2006, 177, 8604–8611.
- Zininga, T.; Ramatsui, L.; Shonhai, A. Heat Shock Proteins as Immunomodulants. Molecules 2018, 23, 2846.
- Asea, A.; Rehli, M.; Kabingu, E.; Boch, J.A.; Bare, O.; Auron, P.E.; Stevenson, M.A.; Calderwood, S.K. Novel signal transduction pathway utilized by extracellular HSP70: Role of toll-like receptor (TLR) 2 and TLR4. J. Biol. Chem. 2002, 277, 15028–15034.
- Lehnardt, S.; Schott, E.; Trimbuch, T.; Laubisch, D.; Krueger, C.; Wulczyn, G.; Nitsch, R.; Weber, J.R. A vicious cycle involving release of heat shock protein 60 from injured cells and activation of toll-like receptor 4 mediates neurodegeneration in the CNS. J. Neurosci. 2008, 28, 2320–2331.
- Fiebich, B.L.; Batista, C.R.A.; Saliba, S.W.; Yousif, N.M.; de Oliveira, A.C.P. Role of Microglia TLRs in Neurodegeneration. Front. Cell Neurosci. 2018, 12, 329.
- Zhang, D.; Sun, L.; Zhu, H.; Wang, L.; Wu, W.; Xie, J.; Gu, J. Microglial LOX-1 reacts with extracellular HSP60 to bridge neuroinflammation and neurotoxicity. Neurochem. Int. 2012, 61, 1021–1035.
- Rosenberger, K.; Dembny, P.; Derkow, K.; Engel, O.; Krüger, C.; Wolf, S.A.; Kettenmann, H.; Schott, E.; Meisel, A.; Lehnardt, S. Intrathecal heat shock protein 60 mediates neurodegeneration and demyelination in the CNS through a TLR4- and MyD88-dependent pathway. Mol. Neurodegener. 2015, 10, 5.
- Giffard, R.G.; Han, R.; Emery, J.F.; Duan, M.; Pittet, J.F. Regulation of apoptotic and inflammatory cell signaling in cerebral ischemia: The complex roles of heat shock protein 70. Anesthesiology 2008, 109, 339–348.
- Yang, C.K.; He, S.D. Heat shock protein 90 regulates necroptosis by modulating multiple signaling effectors. Cell Death Dis. 2016, 7, e2126.
- Mifflin, L.; Ofengeim, D.; Yuan, J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat. Rev. Drug Discov. 2020, 19, 553–571.
- Binder, R.J.; Zhou, Y.J.; Messmer, M.N.; Pawaria, S. CD91-Dependent Modulation of Immune Responses by Heat Shock Proteins: A Role in Autoimmunity. Autoimmune Dis. 2012, 2012, 863041.
- Cohen-Sfady, M.; Nussbaum, G.; Pevsner-Fischer, M.; Mor, F.; Carmi, P.; Zanin-Zhorov, A.; Lider, O.; Cohen, I.R. Heat shock protein 60 activates B cells via the TLR4-MyD88 pathway. J. Immunol. 2005, 175, 3594–3602.
- Murshid, A.; Gong, J.; Calderwood, S.K. The role of heat shock proteins in antigen cross presentation. Front. Immunol. 2012, 3, 63.
- Liu, G.; Zhao, Y. Toll-like receptors and immune regulation: Their direct and indirect modulation on regulatory CD4+ CD25+ T cells. Immunology 2007, 122, 149–156.
- Chen, J.; Graham, S.H.; Zhu, R.L.; Simon, R.P. Stress proteins and tolerance to focal cerebral ischemia. J. Cereb. Blood Flow Metab. 1996, 16, 566–577.
- Lee, J.E.; Kim, Y.J.; Kim, J.Y.; Lee, W.T.; Yenari, M.A.; Giffard, R.G. The 70 kDa heat shock protein suppresses matrix metalloproteinases in astrocytes. Neuroreport 2004, 15, 499–502.
- Zheng, Z.; Kim, J.Y.; Ma, H.; Lee, J.E.; Yenari, M.A. Anti-inflammatory effects of the 70 kDa heat shock protein in experimental stroke. J. Cereb. Blood Flow Metab. 2008, 28, 53–63.
- Sheppard, P.W.; Sun, X.; Khammash, M.; Giffard, R.G. Overexpression of heat shock protein 72 attenuates NF-κB activation using a combination of regulatory mechanisms in microglia. PLoS Comput. Biol. 2014, 10, e1003471.
- Evgen’ev, M.B.; Krasnov, G.S.; Nesterova, I.V.; Garbuz, D.G.; Karpov, V.L.; Morozov, A.V.; Snezhkina, A.V.; Samokhin, A.N.; Sergeev, A.; Kulikov, A.M.; et al. Molecular Mechanisms Underlying Neuroprotective Effect of Intranasal Administration of Human Hsp70 in Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2017, 59, 1415–1426.
- Ferat-Osorio, E.; Sánchez-Anaya, A.; Gutiérrez-Mendoza, M.; Boscó-Gárate, I.; Wong-Baeza, I.; Pastelin-Palacios, R.; Pedraza-Alva, G.; Bonifaz, L.C.; Cortés-Reynosa, P.; Pérez-Salazar, E.; et al. Heat shock protein 70 down-regulates the production of toll-like receptor-induced pro-inflammatory cytokines by a heat shock factor-1/constitutive heat shock element-binding factor-dependent mechanism. J. Inflamm. 2014, 11, 19.
- Kim, N.; Kim, J.Y.; Yenari, M.A. Pharmacological induction of the 70-kDa heat shock protein protects against brain injury. Neuroscience 2015, 284, 912–919.
- Chen, Y.; Wang, B.; Liu, D.; Li, J.J.; Xue, Y.; Sakata, K.; Zhu, L.Q.; Heldt, S.A.; Xu, H.; Liao, F.F. Hsp90 chaperone inhibitor 17-AAG attenuates Aβ-induced synaptic toxicity and memory impairment. J. Neurosci. 2014, 34, 2464–2470.
- Chun, J.N.; Choi, B.; Lee, K.W.; Lee, D.J.; Kang, D.H.; Lee, J.Y.; Song, I.S.; Kim, H.I.; Lee, S.H.; Kim, H.S.; et al. Cytosolic Hsp60 is involved in the NF-kappaB-dependent survival of cancer cells via IKK regulation. PLoS ONE 2010, 5, e9422.
- Lewis, J.; Devin, A.; Miller, A.; Lin, Y.; Rodriguez, Y.; Neckers, L.; Liu, Z.G. Disruption of hsp90 function results in degradation of the death domain kinase, receptor-interacting protein (RIP), and blockage of tumor necrosis factor-induced nuclear factor-kappaB activation. J. Biol. Chem. 2000, 275, 10519–10526.
- Nakamura, H.; Minegishi, H. HSP60 as a drug target. Curr. Pharm. Des. 2013, 19, 441–451.
- Zuo, Y.; Wang, J.; Liao, F.; Yan, X.; Li, J.; Huang, L.; Liu, F. Inhibition of Heat Shock Protein 90 by 17-AAG Reduces Inflammation via P2X7 Receptor/NLRP3 Inflammasome Pathway and Increases Neurogenesis after Subarachnoid Hemorrhage in Mice. Front. Mol. Neurosci. 2018, 11, 401.
- Cappello, F.; Marino Gammazza, A.; Palumbo Piccionello, A.; Campanella, C.; Pace, A.; Conway de Macario, E.; Macario, A.J.L. Hsp60 chaperonopathies and chaperonotherapy: Targets and agents. Expert. Opin. Ther. Targets 2014, 18, 185–208.
- Peterson, L.B.; Blagg, B.S. To fold or not to fold: Modulation and consequences of Hsp90 inhibition. Future Med. Chem. 2009, 1, 267–283.
- Theodoraki, M.A.; Caplan, A.J. Quality control and fate determination of Hsp90 client proteins. Biochim. Biophys. Acta 2012, 1823, 683–688.
- Luo, W.; Sun, W.; Taldone, T.; Rodina, A.; Chiosis, G. Heat shock protein 90 in neurodegenerative diseases. Mol. Neurodegener. 2010, 5, 24.
- Radons, J. The human HSP70 family of chaperones: Where do we stand? Cell Stress Chaperones 2016, 21, 379–404.
- Banjara, M.; Ghosh, C. Sterile Neuroinflammation and Strategies for Therapeutic Intervention. Int. J. Inflamm. 2017, 8385961.
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153.
- Carson, M.J.; Thrash, J.C.; Walter, B. The cellular response in neuroinflammation: The role of leukocytes, microglia and astrocytes in neuronal death and survival. Clin. Neurosci. Res. 2006, 6, 237–245.
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory Response in the CNS: Friend or Foe? Mol. Neurobiol. 2017, 54, 8071–8089.
- Penke, B.; Bogár, F.; Crul, T.; Sántha, M.; Tóth, M.E.; Vígh, L. Heat Shock Proteins and Autophagy Pathways in Neuroprotection: From Molecular Bases to Pharmacological Interventions. Int. J. Mol. Sci. 2018, 19, 325.
- Campanella, C.; Bucchieri, F.; Merendino, A.M.; Fucarino, A.; Burgio, G.; Corona, D.F.; Barbieri, G.; David, S.; Farina, F.; Zummo, G.; et al. The odyssey of Hsp60 from tumor cells to other destinations includes plasma membrane-associated stages and Golgi and exosomal protein-trafficking modalities. PLoS ONE 2012, 7, e42008.
- Broquet, A.H.; Thomas, G.; Masliah, J.; Trugnan, G.; Bachelet, M. Expression of the molecular chaperone Hsp70 in detergent-resistant microdomains correlates with its membrane delivery and release. J. Biol. Chem. 2003, 278, 21601–21606.
- Mambula, S.S.; Stevenson, M.A.; Ogawa, K.; Calderwood, S.K. Mechanisms for Hsp70 secretion: Crossing membranes without a leader. Methods 2007, 43, 168–175.
- Kirkegaard, T.; Roth, A.G.; Petersen, N.H.; Mahalka, A.K.; Olsen, O.D.; Moilanen, I.; Zylicz, A.; Knudsen, J.; Sandhoff, K.; Arenz, C.; et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature 2010, 463, 549–553.
- Lauwers, E.; Wang, Y.C.; Gallardo, R.; Van der Kant, R.; Michiels, E.; Swerts, J.; Baatsen, P.; Zaiter, S.S.; McAlpine, S.R.; Gounko, N.V.; et al. Hsp90 Mediates Membrane Deformation and Exosome Release. Mol. Cell 2018, 71, 689–702.
- Lachenal, G.; Pernet-Gallay, K.; Chivet, M.; Hemming, F.J.; Belly, A.; Bodon, G.; Blot, B.; Haase, G.; Goldberg, Y.; Sadoul, R. Release of exosomes from differentiated neurons and its regulation by synaptic glutamatergic activity. Mol. Cell Neurosci. 2011, 46, 409–418.
- Sun, Y.; Zheng, J.; Xu, Y.; Zhang, X. Paraquat-induced inflammatory response of microglia through HSP60/TLR4 signaling. Hum. Exp. Toxicol. 2018, 37, 1161–1168.
- Marino Gammazza, A.; Caruso Bavisotto, C.; Barone, R.; de Macario, E.C.; Macario, A.J.L. Alzheimer’s disease and molecular chaperones: Current knowledge and the future of chaperonotherapy. Curr. Pharm. Des. 2016, 22, 4040–4049.
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70.
- Mangione, M.R.; Vilasi, S.; Marino, C.; Librizzi, F.; Canale, C.; Spigolon, D.; Bucchieri, F.; Fucarino, A.; Passantino, R.; Cappello, F.; et al. Hsp60, amateur chaperone in amyloid-beta fibrillogenesis. Biochim. Biophys. Acta 2016, 1860, 2474–2483.
- Cappello, F.; Angileri, F.; Conway de Macario, E.; Macario, A.J.L. Chaperonopathies and chaperonotherapy. Hsp60 as therapeutic target in cancer: Potential benefits and risks. Curr. Pharm. Des. 2013, 19, 452–457.
- Cappello, F.; Conway de Macario, E.; Marino Gammazza, A.; Bonaventura, G.; Carini, F.; Czarnecka, A.M.; Farina, F.; Zummo, G.; Macario, A.J.L. Hsp60 and human aging: Les liaisons dangereuses. Front. Biosci. 2013, 18, 626–637.
- Nemirovsky, A.; Fisher, Y.; Baron, R.; Cohen, I.R.; Monsonego, A. Amyloid beta-HSP60 peptide conjugate vaccine treats a mouse model of Alzheimer’s disease. Vaccine 2011, 29, 4043–4050.
- Arispe, N.; De Maio, A. Memory Loss and the Onset of Alzheimer’s Disease Could Be under the Control of Extracellular Heat Shock Proteins. J. Alzheimers Dis. 2018, 63, 927–934.
- De Mena, L.; Chhangani, D.; Fernandez-Funez, P.; Rincon-Limas, D.E. secHsp70 as a tool to approach amyloid-β42 and other extracellular amyloids. Fly 2017, 11, 179–184.
- Kakimura, J.; Kitamura, Y.; Takata, K.; Umeki, M.; Suzuki, S.; Shibagaki, K.; Taniguchi, T.; Nomura, Y.; Gebicke-Haerter, P.J.; Smith, M.A.; et al. Microglial activation and amyloid-beta clearance induced by exogenous heat-shock proteins. FASEB J. 2002, 16, 601–603.
- Maciejewski, A.; Ostapchenko, V.G.; Beraldo, F.H.; Prado, V.F.; Prado, M.A.; Choy, W.Y. Domains of STIP1 responsible for regulating PrPC-dependent amyloid-β oligomer toxicity. Biochem. J. 2016, 473, 2119–2130.
- Wirdefeldt, K.; Adami, H.O.; Cole, P.; Trichopoulos, D.; Mandel, J. Epidemiology and etiology of Parkinson’s disease: A review of the evidence. Eur. J. Epidemiol. 2011, 26, S1–S58.
- Kim, S.D.; Allen, N.E.; Canning, C.G.; Fung, V.S.C. Parkinson disease. Nat. Rev. Dis. Primers 2018, 159, 173–193.
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473.
- Cheng, M.Y.; Hartl, F.U.; Martin, J.; Pollock, R.A.; Kalousek, F.; Neupert, W.; Hallberg, E.M.; Hallberg, R.L.; Horwich, A.L. Mitochondrial heat-shock protein hsp60 is essential for assembly of proteins imported into yeast mitochondria. Nature 1989, 337, 620–625.
- Pemberton, S.; Melki, R. The interaction of Hsc70 protein with fibrillar α-Synuclein and its therapeutic potential in Parkinson’s disease. Commun. Integr. Biol. 2012, 5, 94–95.
- Bohush, A.; Bieganowski, P.; Filipek, A. Hsp90 and Its Co-Chaperones in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 4976.
- Luk, K.C.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M. Interactions between Hsp70 and the hydrophobic core of alpha-synuclein inhibit fibril assembly. Biochemistry 2008, 47, 12614–12625.
- Klucken, J.; Shin, Y.; Masliah, E.; Hyman, B.T.; McLean, P.J. Hsp70 Reduces alpha-Synuclein Aggregation and Toxicity. J. Biol. Chem. 2004, 279, 25497–25502.
- Falsone, S.F.; Kungl, A.J.; Rek, A.; Cappai, R.; Zangger, K. The molecular chaperone Hsp90 modulates intermediate steps of amyloid assembly of the Parkinson-related protein alpha-synuclein. J. Biol. Chem. 2009, 284, 31190–31199.
- Feng, M.; Zhang, L.; Liu, Z.; Zhou, P.; Lu, X. The expression and release of Hsp60 in 6-OHDA induced in vivo and in vitro models of Parkinson’s disease. Neurochem. Res. 2013, 38, 2180–2189.
- Lecours, C.; Bordeleau, M.; Cantin, L.; Parent, M.; Paolo, T.D.; Tremblay, M.È. Microglial Implication in Parkinson’s Disease: Loss of Beneficial Physiological Roles or Gain of Inflammatory Functions? Front. Cell Neurosci. 2018, 12, 282.
- Robberecht, W.; Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264.
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62.
- McAlary, L.; Aquilina, J.A.; Yerbury, J.J. Susceptibility of mutant SOD1 to form a destabilized monomer predicts cellular aggregation and toxicity but not in vitro aggregation propensity. Front. Neurosci. 2016, 10, 499.
- San Gil, R.; Ooi, L.; Yerbury, J.J.; Ecroyd, H. The heat shock response in neurons and astroglia and its role in neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 65.
- Takeuchi, H.; Kobayashi, Y.; Yoshihara, T.; Niwa, J.; Doyu, M.; Ohtsuka, K.; Sobue, G. Hsp70 and Hsp40 improve neurite outgrowth and suppress intracytoplasmic aggregate formation in cultured neuronal cells expressing mutant SOD1. Brain Res. 2002, 949, 11–22.
- Liu, J.; Shinobu, L.A.; Ward, C.M.; Young, D.; Cleveland, D.W. Elevation of the Hsp70 chaperone does not effect toxicity in mouse models of familial amyotrophic lateral sclerosis. J. Neurochem. 2005, 93, 875–882.
- Apolloni, S.; Caputi, F.; Pignataro, A.; Amadio, S.; Fabbrizio, P.; Ammassari-Teule, M.; Volonté, C. Histamine Is an Inducer of the Heat Shock Response in SOD1-G93A Models of ALS. Int. J. Mol. Sci. 2019, 20, 3793.
- Gifondorwa, D.J.; Robinson, M.B.; Hayes, C.D.; Taylor, A.R.; Prevette, D.M.; Oppenheim, R.W.; Caress, J.; Milligan, C.E. Exogenous delivery of heat shock protein 70 increases lifespan in a mouse model of amyotrophic lateral sclerosis. J. Neurosci. 2007, 27, 13173–13180.
- Tytell, M. Release of heat shock proteins (Hsps) and the effects of extracellular Hsps on neural cells and tissues. Int. J. Hyperth. 2005, 21, 445–455.
- Giffard, R.G.; Yenari, M.A. Many mechanisms for hsp70 protection from cerebral ischemia. J. Neurosurg. Anesth. 2004, 16, 53–61.
- Taylor, A.R.; Robinson, M.B.; Gifondorwa, D.J.; Tytell, M.; Milligan, C.E. Regulation of heat shock protein 70 release in astrocytes: Role of signaling kinases. Dev. Neurobiol. 2007, 67, 1815–1829.
- Fontaine, S.N.; Zheng, D.; Sabbagh, J.J.; Martin, M.D.; Chaput, D.; Darling, A.; Trotter, J.H.; Stothert, A.R.; Nordhues, B.A.; Lussier, A.; et al. DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J. 2016, 35, 1537–1549.
- Robinson, M.B.; Taylor, A.R.; Gifondorwa, D.J.; Tytell, M.; Milligan, C.E. Exogenous Hsc70, but not thermal preconditioning, confers protection to motoneurons subjected to oxidative stress. Dev. Neurobiol. 2008, 68, 1–17.