Insulin resistance (IR) is a condition in which insulin action is altered. In metabolic terms, IR represents the inability of a fixed amount of insulin to metabolize a known amount of glucose in an individual as compared to the general population.
- NAFLD
- insulin resistance
- metabolic syndrome
1. Insulin Resistance
Insulin resistance (IR) is closely related to visceral fat and represents the cornerstone of metabolic syndrome (MS), which is defined by the presence of at least three among the following features: abdominal obesity, increased triglycerides levels, reduced HDL cholesterol levels, increased blood pressure, and hyperglycemia [1].
Insulin hormone is produced by the beta cells of the pancreas. Once secreted, it binds to the extracellular domain of its specific receptor, thus causing a conformational change, with the subsequent phosphorylation of specific tyrosine residues on the intracytoplasmic domain of the receptor itself [2]. Then, the activated receptor hooks the so-called insulin receptor substrates (IRS), which are in turn activated by tyrosine-phosphorylation processes. This cascade of events triggers a series of downstream phosphorylation inside the cell, which thus becomes able to mediate the effects of insulin. In detail, IRS molecules become able to activate phosphatidylinositol 3-kinase (PI3K), thus causing the translocation of glucose transporter type-4 (GLUT-4) protein from the cytoplasm to the cytoplasmic membrane. In this way, GLUT-4 is able to capture glucose from the bloodstream and internalize it inside the cell. In addition to the crucial role of enabling intracellular glucose entry, activated PI3K may also trigger anti-lipolytic effects, activation of fatty acid synthase, and glycogen synthase, thus mediating the typical anabolic effects of insulin. Phosphorylated IRS also leads to the activation of the Ras/MAPK pathway, involved in cell survival and stimulation of mitosis [3]. This suggests that the two biochemical cascades activated by insulin, i.e., the regulation of intermediate metabolism and the stimulation of cell growth and proliferation, might be dissociated in their regulation.
The complexity of the insulin metabolic pathway explains why several interferences may affect its biochemical signal. On the other hand, IR may be mediated by alterations at any level of its biochemical signal cascade. IR rising during MS is characterized by the impact of glucose on cellular absorption processes, by the suppression of lipolysis of adipose tissue, and the impairment of vasodilation, whilst action on growth and cytogenesis are not affected [4]. The selective inhibition of insulin action on metabolism and vasodilation might be explained by interference at IRS and PI3K [5].
Genetically induced alterations triggering IR are heterogeneous and may affect the bond between insulin and its receptor, the autophosphorylation process, and the kinase activity of either IRS or PI3K or MAP kinase [6]8]. However, IR during MS is mainly determined by environmental factors due to an improper lifestyle, i.e., a discrepancy between caloric intake and energy consumption. As a consequence, the development of visceral obesity, a risk factor for the onset of both T2DM and cardiovascular (CV) diseases, is the result of this positive energy balance [7][8]. The flow of fatty acids originating from the visceral adipose tissue determines its pathophysiologic effects [9], thus establishing a direct correlation between IR and the amount of visceral adipose tissue mass [10]. In fact, the action of insulin is mainly related to the visceral adipose tissue rather than to the subcutaneous adipose tissue. In this regard, weight loss has been proven to improve insulin sensitivity, as well as its association with changes in the mass of visceral adipose tissue, and of either total or subcutaneous adipose tissue [11][12].
Metabolic regulation of the neuroendocrine axis includes adipose tissue, alongside the central nervous system (CNS) and intestine, through the regulation of insulin sensitivity in the target tissues [13]. Adipocytes store and release fatty acids in the form of triglycerides according to the body’s needs. Target tissues (e.g., the skeletal muscle) may oxidize fatty acids and use them as fuel. However, elevated levels of circulating fatty acids can desensitize target tissues to the actions of insulin.
Adipocytes also secrete other hormones: adipokines. Among them, adiponectin acts as a stimulator of insulin action in the peripheral tissues [14], whilst leptin and resistin inhibit the sensitivity to insulin action [15]. In the presence of visceral obesity, leptin levels increase whilst adiponectin reduces. Moreover, adipocytes secrete chemokines, which are able to enroll macrophages inside the adipose tissue, responsible, in turn, for the increased levels of tumor necrosis factor-alpha (TNF-α) [16]. The latter, as resistin and other pro-inflammatory cytokines such as interleukin-6 (IL-6), enhance the insulin resistance phenomena and correlates with the endothelial dysfunction degree [17].
As aforementioned, the increase in the mass of visceral adipose tissue also determines, besides the imbalance of adipokine levels, an increase in free fatty acids (FFA) levels. In turn, an excess of FFA is able to determine IR through the inhibition of the post-receptor insulin signal. Indeed, the increase in FFA determines the activation of a serine kinase protein able to phosphorylate the serine residues on IRS and PI3K molecules [18]. Then, IRS and PI3K can no longer be phosphorylated at the level of their tyrosine residues and thus cannot be adequately activated, with the subsequent result of a block in the transmission of insulin signal [19]. Similar effects are also caused by elevated levels of TNF-α and IL-6 [20]. The increase in plasma circulating triglycerides also determines an elevated synthesis of plasminogen activator inhibitor-1 (PAI-1) by endothelial cells, supporting the subsequent endothelial dysfunction and the clinical development of arterial hypertension [21]. Therefore, an extremely positive caloric balance due to an unhealthy lifestyle causes IR by the increase in visceral adipose tissue and the consequent release of FFA, TNF-α, and adipokines (Figure 1).
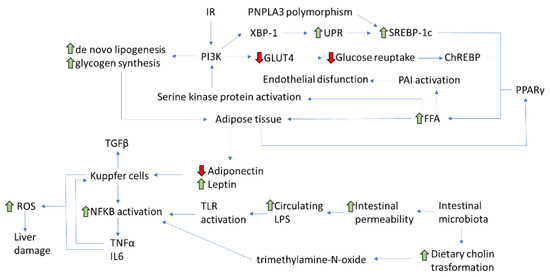
Figure 1. Main pathophysiological pathways involved in NAFLD development (IR: Insulin resistance; PNPLA3: patatin-like phospholipase domain-containing protein 3; XBP: X-box Binging Protein-1; UPR: Unfolded Protein Response; SREBP-:1 sterol regulatory element-binding protein-1; PI3K: phosphatidylinositol 3-kinase; GLUT-4: glucose transporter type-4; ChREBP: carbohydrate-responsive element-binding protein; PAI-1: plasminogen activator inhibitor-1; PPAR-γ: peroxisome proliferator-activated receptor-gamma; FFA: free fatty acids; TGF-β1: transforming growth factor-beta 1; TNF-α: tumor necrosis factor-alpha; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; TLR: toll-like receptors; LPS: lipopolysaccharide; IL6: interleukin -6; ROS: Reactive Oxygen Species).
2. Non-Alcoholic Fatty Liver Disease (NAFLD) and Insulin Resistance
NAFLD is characterized by the accumulation of triglycerides in over 5% of hepatocytes in absence of alcohol consumption.
NAFLD clinical presentations is heterogeneous including simple steatosis or non-alcoholic steatoepatitis (NASH), characterized by necro-inflammatory activity. Both conditions may either regress or progress to liver cirrhosis and its complications (e.g. liver failure, portal hypertension, epatocelluar carcinoma). At present, NAFLD has became the most frequent cause of chronic liver disease and its growing prevalence is related to overeating, overweight/obesity and sedentary lifestyle. NAFLD, indeed, could be both cause and effect of type 2 diabetes and metabolic syndrome. In additon, NAFLD etiology could involve genetic factors (gene polymorphisms: PNPLA3 and TM6SF2) and could also affect lean and non-diabetic people.
IR plays a key role in the development of NAFLD, as it causes an increase in hepatic lipogenesis and an inhibition of adipose tissue lipolysis, with a subsequent elevated flow of fatty acids in the liver [22][23]. Fat accumulates within the hepatocytes mainly as triglycerides deriving from the esterification of glycerol and FFAs [24][25]. The hepatic accumulation of triglycerides does not represent itself as a hepatotoxic event, rather as a defense mechanism able to balance the excess FFAs in the plasma [26]. However, other bioactive intermediates, such as ceramides and diacylglycerol (DAG), can induce lipotoxicity, resulting in inflammation, necrosis, and liver fibrosis. NAFLD progresses to NASH when the mechanisms protecting hepatocytes from lipotoxicity are depleted. This induces necrosis and secondary repair phenomena mediated by hepatic stellate cells, with the deposition of scar collagen tissue, hence the development and progression of fibrosis [27].
As already described, NAFLD patients display a de novo hepatic lipogenesis higher than healthy controls, which is neither suppressed in fasting nor with higher FFA plasma levels [28]. Hepatic lipogenesis during IR can be further induced by the activation of transcription factors, such as the sterol regulatory element-binding protein-1 (SREBP-1), the carbohydrate-responsive element-binding protein (ChREBP), and the peroxisome proliferator-activated receptor-gamma (PPAR-γ) [29]. SREBP-1 is a transcription factor present in different isoforms: SREBP-1c, which regulates de novo lipogenesis and is stimulated by insulin, and SREBP-2, instead involved in cellular cholesterol homeostasis [30]. ChREBP is activated by glucose and induces lipogenesis, but it also provides more substrates for the synthesis of both triglycerides and FFA. Among insulin receptors, IRS-2, when activated, can act as a regulator of SREBP-1c, thus affecting de novo lipogenesis [31]. Under IR conditions, IRS-2 is downregulated, with overexpression of SREBP-1c resulting in a stimulation of lipogenesis [32]. At the same time, FFA beta-oxidation is inhibited, which further favors the hepatic accumulation of lipids [33]. In addition, insulin has a powerful suppression action on adipose tissue lipolysis, usually compromised during IR, resulting in an increase in the flow of FFA to the liver [34]. Once accumulated in the liver, FFAs induce alterations in the insulin signaling pathways through the activation of serine kinase and contribute to the worsening of the systemic state of IR [35]. Conversely, NAFLD itself contributes to the development of IR, leading to the establishment of a vicious circle. In fact, in NAFLD patients, genetic and environmental factors can interfere with the insulin post-receptor signal cascade through the phosphorylation of serine residues on insulin [36], the activation of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and the Suppressor Of Cytokine Signaling-3 (SOCS-3) [37]. Therefore, the relationship between IR and NAFLD seems bidirectional. Indeed, the complex network of events involved in the development of NASH may determine an increase in hepatic IR up-regulating SOCS-3 which, through the interference with IRS-1, determines a reduced insulin signal transmission [38]. Therefore, it seems likely that an improvement in IR degree could help to reduce NAFLD progression and should thus be considered among the main therapeutic targets.
This entry is adapted from the peer-reviewed paper 10.3390/pr9010135
References
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C., Jr.; et al. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645.
- Sonksen, P.; Sonksen, J. Insulin: Understanding its action in health and disease. Br. J. Anaesth. 2000, 85, 69–79.
- Posner, B.I. Insulin Signalling: The Inside Story. Can. J. Diabetes 2017, 41, 108–113.
- Goetze, S.; Kim, S.; Xi, X.P.; Graf, K.; Yang, D.C.; Fleck, E.; Meehan, W.P.; Hsueh, W.A.; Law, R.E. Troglitazone inhibits mitogenic signaling by insulin in vascular smooth muscle cells. J. Cardiovasc. Pharmacol. 2000, 35, 749–757.
- Kim, Y.B.; Nikoulina, S.E.; Ciaraldi, T.P.; Henry, R.R.; Kahn, B.B. Normal insulin-dependent activation of Akt/protein kinase B, with diminished activation of phosphoinositide 3-kinase, in muscle in type 2 diabetes. J. Clin. Investig. 1999, 104, 733–741.
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin resistance: Review of the underlying molecular mechanisms. J. Cell Physiol. 2019, 234, 8152–8161.
- Kachur, S.; Lavie, C.J.; de Schutter, A.; Milani, R.V.; Ventura, H.O. Obesity and cardiovascular diseases. Minerva Med. 2017, 108, 212–228.
- Cozzolino, D.; Sessa, G.; Salvatore, T.; Sasso, F.C.; Giugliano, D.; Lefebvre, P.J.; Torella, R. The involvement of the opioid system in human obesity: A study in normal weight relatives of obese people. J. Clin. Endocrinol. Metab. 1996, 81, 713–718.
- Montague, C.T.; O’Rahilly, S. The perils of portliness: Causes and consequences of visceral adiposity. Diabetes 2000, 49, 883–888.
- Banerji, M.A.; Lebowitz, J.; Chaiken, R.L.; Gordon, D.; Kral, J.G.; Lebovitz, H.E. Relationship of visceral adipose tissue and glucose disposal is independent of sex in black NIDDM subjects. Am. J. Physiol. 1997, 273, E425–E432.
- Goodpaster, B.H.; Kelley, D.E.; Wing, R.R.; Meier, A.; Thaete, F.L. Effects of weight loss on regional fat distribution and insulin sensitivity in obesity. Diabetes 1999, 48, 839–847.
- Banerji, M.A.; Buckley, M.C.; Chaiken, R.L.; Gordon, D.; Lebovitz, H.E.; Kral, J.G. Liver fat, serum triglycerides and visceral adipose tissue in insulin-sensitive and insulin-resistant black men with NIDDM. Int. J. Obes. Relat. Metab. Disord. 1995, 19, 846–850.
- Zac-Varghese, S.; Tan, T.; Bloom, S.R. Hormonal interactions between gut and brain. Discov. Med. 2010, 10, 543–545.
- Ahima, R.S.; Lazar, M.A. Adipokines and the peripheral and neural control of energy balance. Mol. Endocrinol. 2008, 22, 1023–1031.
- Park, H.K.; Kwak, M.K.; Kim, H.J.; Ahima, R.S. Linking resistin, inflammation, and cardiometabolic diseases. Korean J. Intern. Med. 2017, 32, 239–247.
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246.
- Esposito, K.; Ciotola, M.; Sasso, F.C.; Cozzolino, D.; Saccomanno, F.; Assaloni, R.; Ceriello, A.; Giugliano, D. Effect of a single high-fat meal on endothelial function in patients with the metabolic syndrome: Role of tumor necrosis factor-alpha. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 274–279.
- Dresner, A.; Laurent, D.; Marcucci, M.; Griffin, M.E.; Dufour, S.; Cline, G.W.; Slezak, L.A.; Andersen, D.K.; Hundal, R.S.; Rothman, D.L.; et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J. Clin. Investig. 1999, 103, 253–259.
- Hotamisligil, G.S.; Spiegelman, B.M. Tumor necrosis factor alpha: A key component of the obesity-diabetes link. Diabetes 1994, 43, 1271–1278.
- Loskutoff, D.J.; Samad, F. The adipocyte and hemostatic balance in obesity: Studies of PAI-1. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1–6.
- Bugianesi, E.; Moscatiello, S.; Ciaravella, M.F.; Marchesini, G. Insulin resistance in nonalcoholic fatty liver disease. Curr. Pharm Des. 2010, 16, 1941–1951.
- Watt, M.J.; Miotto, P.M.; De Nardo, W.; Montgomery, M.K. The Liver as an Endocrine Organ-Linking NAFLD and Insulin Resistance. Endocr. Rev. 2019, 40, 1367–1393.
- Jacome-Sosa, M.M.; Parks, E.J. Fatty acid sources and their fluxes as they contribute to plasma triglyceride concentrations and fatty liver in humans. Curr. Opin. Lipidol. 2014, 25, 213–220.
- Musso, G.; Gambino, R.; Cassader, M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog. Lipid Res. 2013, 52, 175–191.
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374.
- Jou, J.; Choi, S.S.; Diehl, A.M. Mechanisms of disease progression in nonalcoholic fatty liver disease. Semin. Liver Dis. 2008, 28, 370–379.
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735.
- George, J.; Liddle, C. Nonalcoholic fatty liver disease: Pathogenesis and potential for nuclear receptors as therapeutic targets. Mol. Pharm. 2008, 5, 49–59.
- Schultz, J.R.; Tu, H.; Luk, A.; Repa, J.J.; Medina, J.C.; Li, L.; Schwendner, S.; Wang, S.; Thoolen, M.; Mangelsdorf, D.J.; et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000, 14, 2831–2838.
- Schreuder, T.C.; Verwer, B.J.; van Nieuwkerk, C.M.; Mulder, C.J. Nonalcoholic fatty liver disease: An overview of current insights in pathogenesis, diagnosis and treatment. World J. Gastroenterol. 2008, 14, 2474–2486.
- Stefan, N.; Kantartzis, K.; Häring, H.U. Causes and metabolic consequences of Fatty liver. Endocr. Rev. 2008, 29, 939–960.
- Postic, C.; Girard, J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: Lessons from genetically engineered mice. J. Clin. Investig. 2008, 118, 829–838.
- Lewis, G.F.; Carpentier, A.; Adeli, K.; Giacca, A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr. Rev. 2002, 23, 201–229.
- Hotamisligil, G.S. Role of endoplasmic reticulum stress and c-Jun NH2-terminal kinase pathways in inflammation and origin of obesity and diabetes. Diabetes 2005, 54, S73–S78.
- Sabio, G.; Das, M.; Mora, A.; Zhang, Z.; Jun, J.Y.; Ko, H.J.; Barrett, T.; Kim, J.K.; Davis, R.J. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 2008, 322, 1539–1543.
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96.
- Torisu, T.; Sato, N.; Yoshiga, D.; Kobayashi, T.; Yoshioka, T.; Mori, H.; Iida, M.; Yoshimura, A. The dual function of hepatic SOCS3 in insulin resistance in vivo. Genes Cells 2007, 12, 143–154.