In recent years, lipid metabolism has gained greater attention in several diseases including cancer. Dysregulation of fatty acid metabolism is a key component in breast cancer malignant transformation. In particular, de novo lipogenesis provides the substrate required by the proliferating tumor cells to maintain their membrane composition and energetic functions during enhanced growth. However, it appears that not all breast cancer subtypes depend on de novo lipogenesis for fatty acid replenishment. Indeed, while breast cancer luminal subtypes rely on de novo lipogenesis, the basal-like receptor-negative subtype overexpresses genes involved in the utilization of exogenous-derived fatty acids, in the synthesis of triacylglycerols and lipid droplets, and fatty acid oxidation. These metabolic differences are specifically associated with genomic and proteomic changes that can perturb lipogenic enzymes and related pathways. This behavior is further supported by the observation that breast cancer patients can be stratified according to their molecular profiles. Moreover, the discovery that extracellular vesicles act as a vehicle of metabolic enzymes and oncometabolites may provide the opportunity to noninvasively define tumor metabolic signature.
- breast cancer
- de novo lipogenesis
1. Introduction
Breast cancer is the most common cancer in female individuals. In 2020, breast, lung, and colorectal accounted for 50% of all new diagnoses of female cancers, and breast cancer alone accounts for 30% [1]. Breast cancer is a highly heterogeneous disease characterized by the presence of different subtypes with variable clinical outcomes. With the growing amount of data generated by proteogenomic studies, researchers have been able to define a detailed molecular landscape of each subtype, thus providing a detailed picture of genomic alterations and altered functional networks [2][3][4].
Transcriptional profiling studies have defined four major breast cancer subtypes: luminal A, luminal B, HER2-enriched, and basal-like [5]. Luminal breast cancers, which include luminal subtypes A and B, are characterized by estrogen receptor (ER) and/or progesterone (PR) expression and confers a more favorable prognosis in part due to reactivity to anti-hormone therapies [6]. In contrast, baseline-type breast cancer such as ER negative, PR negative, and HER2 negative (triple negative breast cancer, TNBC) has the highest recurrence rate and worst overall survival rate among all breast cancer subtypes [6]. As a consequence of this different genomic and proteomic background, breast cancers can also be classified in terms of altered metabolic pathways and metabolite levels. For instance, from the analysis of 228 non-treated breast cancer patients, three significantly different metabolic clusters (Mc1, Mc2, and Mc3) have emerged. Mc1 is characterized by the highest levels of glycerophosphocholine and phosphocholine, Mc2 by the highest levels of glucose, and Mc3 by the highest levels of lactate and alanine [7].
This metabolic phenotyping results in enhanced nutrient uptake to support the metabolic demand of tumor cells, but also to provide cells with metabolic intermediates, named oncometabolites, which sustain specific cellular processes [8][9]. For instance, increased lactate production is a characteristic of ER− tumors due to the increased rate of glycolysis and over-expression of lactate dehydrogenases [10].
In this scenario, lipid metabolic reprogramming is a hallmark in cancer [10][11]. Cancer cells exhibit a “lipogenic phenotype” characterized by exacerbated levels of fatty acid biogenesis, even in the presence of abundant circulating exogenous fatty acids and reflected in the overexpression and increased activity of lipogenic enzymes [11][12][13][14]. Fatty acids or derived lipid species are then critical for lipid synthesis and membrane structures, protein modification and localization functions, and receptor localization and signaling of major oncogenic signaling pathways [15].
Accordingly, the uptake of preformed fatty acids may also be an important mechanism of cancer lipid acquisition. Several factors including genetic mutations and metabolic conditions of oxygen and nutrient deprivation can drive this metabolic adaptation [16]. In the context of breast cancer, while the luminal subtype relies on de novo lipogenesis (DNL) (or de novo fatty acid synthesis) as a source for biomass and energy requirements, basal-like TNBC cells overexpress genes involved in the utilization of exogenous fatty acids and triacylglycerol synthesis [17]. Lipid transport and uptake are indeed important and under-appreciated aspects of lipid metabolism in cancer [18]. This aspect links lipid metabolism with the activation of particular tumor cellular processes such as the epithelial–mesenchymal transition (EMT) that is thought to contribute to cancer progression and chemoresistance [19][20][21].
These findings highlight the role of lipid metabolism in breast cancer pathophysiology and the tight correlation with multiple cellular processes. Here, we review this functional crosstalk focusing on metabolic pathways related to DNL.
2. Lipogenesis in Cancer
In normal tissues, glucose represents the main source of acetyl-CoA for lipid synthesis, a process known as DNL (Figure 1).
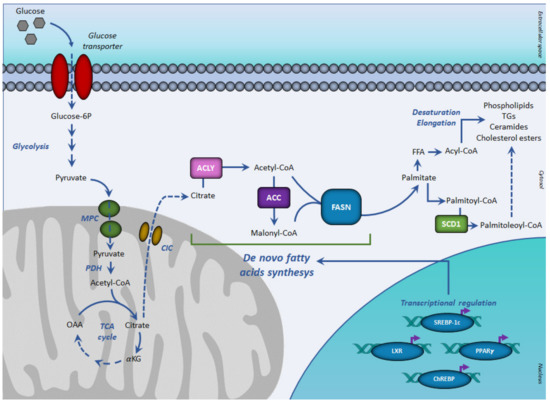
Figure 1. De novo lipogenesis and transcriptional regulation. In aerobic conditions, glucose-derived pyruvate fuels, in the form of acetyl-CoA, the tricarboxylic acid cycle, to form citrate. Once exported in the cytosol, citrate generates acetyl-CoA for fatty acid synthesis, mediated by sequential reactions of acetyl-CoA carboxylase and fatty acid synthase. The resulting palmitoyl-CoA is used for the synthesis of complex lipids. The de novo fatty acids synthesis is regulated at the transcriptional level by the SREBP-1c, PPARγ, ChREBP, and LXR receptor family. Abbreviations: ACC, acetyl-CoA carboxylase; ACLY, ATP citrate lyase; ChREBP, carbohydrate-responsive element-binding protein; CIC, citrate carrier; FASN, fatty acid synthase; FFA, free (non-esterified) fatty acid; αKG, α-ketoglutarate; LXR, liver X receptor; MPC, mitochondrial pyruvate carrier; OAA, oxaloacetate; PPARγ, peroxisome proliferator-activated receptor-γ; PDH, pyruvate dehydrogenase; SCD1, stearoyl-CoA desaturase; SREBP-1c, sterol regulatory element-binding proteins-1c; TCA, tricarboxylic acid cycle.
In this case, pyruvate, the glycolytic end-product, enters mitochondria where it is converted in acetyl-CoA by the pyruvate dehydrogenase complex. In hypoxia, a common feature of solid tumors, a metabolic reprogramming accounts for a reduction in the mitochondrial conversion of pyruvate into acetyl-CoA that normally feeds the tricarboxylic acid cycle (TCA) to produce citrate. As a result, glycolysis-derived pyruvate is primarily diverted to lactate rather than to mitochondrial oxidative phosphorylation [22]. This metabolic reprogramming allows the cancer cells to rapidly obtain ATP and glycolytic intermediates to support anabolic demand.
Cancer cells adopt different metabolic adaptations for fatty acid synthesis, mainly relying on glutamine or acetate as alternative substrates [22]. The reductive carboxylation of glutamine-derived α-ketoglutarate, through the isocitrate dehydrogenase-1 (IDH1)-dependent pathway represents, for different cancers, the main pathway to synthesize acetyl-CoA for lipid synthesis [23]. TNBC cells often exhibit glutamine-dependent phenotype upregulating both glutamine uptake and glutamine-related enzymes [24][25]. Consequently, TNBC may be more susceptible to glutamine-targeting therapeutics compared to luminal types [25].
Acetate can be derived from a range of sources including extracellular acetate, histone deacetylation, and the recently identified conversion of pyruvate into acetate by thiamine-dependent keto acid dehydrogenases as well as a reactive oxygen species-coupled reaction [26].
In hypoxic conditions, breast cancers increase acetate uptake [27][28][29][30] and upregulate enzymes converting acetate to acetyl-CoA such as acetyl-CoA synthetases (ACSS). Indeed, acyl-CoA synthetase short-chain family member 2 (ACSS2) was found to be upregulated in hypoxic breast cells [27][30].
Hypoxic cells can also rely on the uptake of fatty acids to compensate for reduced glucose-based DNL [26][31]. Breast cancers have increased expression of fatty acid-binding proteins (FABP3, FABP7 or FABP4) and CD36 [32][33], which are involved in the uptake and subcellular trafficking of fatty acids [31][34][35]. Thus, in breast cancer cells, lipid accumulation in the form of lipid droplets relies on a FABP-dependent fatty acid uptake, while DNL resulted in being repressed. Indeed, CD36 inhibition impaired angiogenesis as well as migration and invasion of breast cancer cell lines [36][37].
3. Key Enzymes of De Novo Lipogenesis (DNL)
Cell requirement for fatty acids is normally met by the utilization of dietary fatty acids. The amount of fatty acids synthetizes by DNL is of minor importance in most human tissues, except for the liver, mammary gland, and to a lesser extent, adipose tissue [38]. However, the rate of DNL and the expression of several lipogenic enzymes are increased in various cancer types.
DNL is a metabolic process by which pyruvate, mainly derived from carbohydrate sources is converted into fatty acids (Figure 1) [38].
In aerobic conditions, pyruvate, the end product of glycolysis is transformed in acetyl-CoA, which enters the TCA cycle by condensing with oxaloacetate to form citrate. When the energetic charge is high such as after a meal rich in carbohydrates, citrate can be transported from the mitochondria into the cytosol, where fatty acid synthesis occurs. Citrate efflux into the cytosol is catalyzed by the citrate carrier (CIC), an intrinsic protein of the inner mitochondrial membrane, which catalyzes an electroneutral exchange of citrate plus a proton with malate. In the cytosol, glucose-derived citrate is converted into oxaloacetate and acetyl-CoA by the ATP citrate lyase (ACLY). The obtained acetyl-CoA is required for lipid synthesis during membrane biogenesis as well as for histone acetylation reactions to regulate the expression of certain proteins in aberrantly proliferating cancer cells.
Key enzymes of DNL are acetyl-CoA carboxylase (ACC) and the multi enzymatic complex fatty acid synthase (FASN).
ACC represents the rate-limiting enzyme of DNL, catalyzing the irreversible carboxylation of acetyl-CoA into malonyl-CoA. The reaction requires biotin and ATP. In humans and mammals, there are two isoforms of ACCs: ACC1 (or ACC-α) with 265 kDa and ACC2 (or ACC-β) with 280 kDa. ACC1 presents in the cytosol with a lipogenic role, so is particularly expressed in lipogenic tissues and ACC2 is mainly associated with mitochondria in oxidative tissues [39][40][41]. Therefore, ACC1 is enriched in lipogenic tissues such as the liver, adipose, and lactating mammary gland, where it catalyzes the biosynthesis of long-chain fatty acids. In contrast, ACC2 is highly expressed in oxidative tissues such as skeletal muscle and heart, where it regulates fatty acid β-oxidation.
Cytosolic malonyl-CoA, produced by ACC, can be used for fatty acid biosynthesis. The reaction is catalyzed by FASN. After priming with acetyl-CoA, FASN uses malonyl-CoA as a carbon donor and NADPH as a reduced cofactor to produce palmitoyl-CoA.
By furnishing malonyl-CoA, ACC not only plays a key role in DNL, but also regulates mitochondrial fatty acid β-oxidation, considering that malonyl-CoA is an inhibitor of carnitine palmitoyl-transferase-1, the key enzyme of this metabolic process.
The de novo synthesized fatty acids can be used for the synthesis of complex lipids such as phospholipids, ceramides, cholesterol esters, and triacylglycerols and thereby play a major role in membrane structure, cell signaling, and energy storage. Following DNL, the enzyme stearoyl-CoA desaturase (SCD) catalyzes the introduction of the first double bond in the cis-delta 9 position of saturated fatty acyl-CoA giving monounsaturated fatty acids, which are preferentially transformed into triacylglycerols for storage [41]. Two isoforms of SCD have been reported in human cells, SCD1 and SCD5 [42]. Both isoforms are overexpressed in luminal cancer models compared to the TNBC subtypes [43].
4. Transcription Factors Regulating De Novo Lipogenesis (DNL)
DNL is a highly regulated metabolic pathway. Having common features at their promoter regions, lipogenic genes are coordinately regulated at the transcription level. The transcription factors sterol regulatory element-binding protein-1c (SREBP-1c), upstream stimulatory factor (USF), peroxisome-proliferation-activated receptors (PPARs), carbohydrate response element-binding protein (ChREBP), and liver X receptors (LXRs) play critical roles in regulating this process (Figure 1).
SREBPs represent the master transcriptional factors regulating DNL. SREBPs are members of the basic helix-loop-helix (bHLH)-leucine zipper transcription factors and can be classified into three types: SREBP-2, SREBP-1a, and SREBP-1c. Whereas SREBP-2 preferentially regulates genes involved in cholesterol metabolism, SREBP-1 regulates fatty acid synthesis enzymes. Expressions of ACC, FASN, and SCD1 are under the control of SREBP-1c [44]. Moreover, SREBP-1c activates the expression of CIC both in hepatocytes [45] and in the mammary epithelium [46] and SREBP-1 overexpression increases the CIC transcript and protein levels. Moreover, SREBP-1 upregulates ACLY at the mRNA level via Akt signaling [47].
Although SREBP-1 plays a pivotal role in regulating lipogenic gene expression, it is not the only one. In vitro studies have demonstrated that insulin effect on FASN promoter also requires the presence of the upstream stimulatory factors (USFs). USFs are bHLH-leucine zipper transcription factors able to bind the CANNTG sequence present in the promoter region of FASN. The effects of SREBP-1 and USFs on FASN are independent and additive [48].
Lipogenic enzyme transcription may also be regulated by ChREBP [49], a glucose-regulated bHLH transcription factor. In response to increased glucose levels, ChREBP undergoes dephosphorylation steps that allow translocation from the cytoplasm to the nucleus where, in association with its binding partner Max-like (MLX) interacting protein, it binds carbohydrate response elements of lipogenic genes [50][51][52].
LXRs are members of the nuclear receptor superfamily that heterodimerize with retinoid X receptor (RXR) [53]. Two isoforms of LXRs have been identified, LXRα and LXRβ [53][54]. It has been reported that LXRs perform an important role in the regulation of fatty acid synthesis. LXRs can activate lipogenic enzymes directly or by SREBP-1c. FASN is transcriptionally regulated by both LXRα and LXRβ [54][55][56].
PPARs are members of the superfamily of nuclear hormone receptors that function as ligand-dependent transcription factors. Upon ligand activation, they regulate the expression of genes containing a specific response element, called the PPAR-responsive element (PPRE), which consists of a hexameric nucleotide direct repeat of the recognition motif (TGACCT) spaced by one nucleotide (DR-1). Three subtypes of PPARs termed α, δ (or β), and γ, have been identified [57][58]. These receptors heterodimerize with the retinoid X receptor (RXR) and alter the transcription of target genes after binding to PPRE.
Although PPARγ is considered to be the master regulator of adipocyte differentiation, an increase in PPARγ expression has been associated with accumulation in hepatic triacylglycerols. A study reports that PPARγ is capable of inducing lipid accumulation in hepatocytes in which an increase in SREBP-1 as well as ACC and FASN expression is also measured. These data suggest that PPARγ may play a role in stimulating lipogenesis [57][58][59]. Heterozygous PPARγ mutant mice exhibit smaller fat stores upon a high-fat diet [60][61]. Recently, it has been reported that the overexpression of PPARα/RXRα and PPARγ/RXRα heterodimers enhances CIC promoter activity in BRL-3A and 3T3-L1 cells, respectively [45].
5. Role of DNL Enzymes in Breast Cancer
Oncogenic signaling has been reported to increase DNL in order to prepare the cell for invasion and metastasis. However, it appears that not all breast cancer subtypes depend on DNL for fatty acid supply. Indeed, while the luminal subtypes rely on DNL, the TNBC subtype overexpresses genes involved in the utilization of exogenous-derived fatty acids, in the synthesis of triacylglycerols and lipid droplets, and fatty acid oxidation (Figure 2).
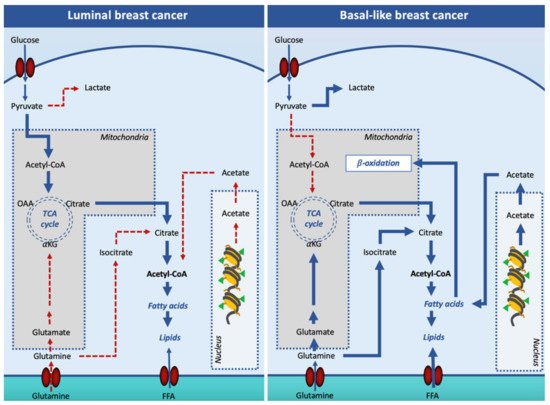
Figure 2. Lipogenesis in luminal and basal-like breast cancer cells. In luminal breast cancer cells, glucose-derived acetyl-CoA is the main source of citrate for the cytosolic synthesis of lipids. In basal-like, pyruvate is mainly converted into lactate in the cytosol, and other substrates such as glutamine and acetate are used to support cell lipid synthesis. Additionally, basal-like breast cancer cells increase free fatty acid entry in the cell to fulfill the β-oxidation pathway. Solid arrows signify the main reaction processes and dotted arrows signify processes with a minor relevance. Abbreviations: FFA, free (non-esterified) fatty acid; OAA, oxaloacetate; αKG, α-ketoglutarate; TCA, tricarboxylic acid cycle.
To support the high demand of acetyl-CoA for the increased DNL, luminal breast cancer cells increase glucose entry and glycolytic flux [62]. By transporting citrate into the cytosol, CIC plays an important role in DNL. Thus, CIC inhibition can potentially limit cancer cell proliferation. Indeed, inhibition of CIC activity by BTA was reported to reduce breast xenograft tumor growth [63].
Changes in ACLY expression have been found in diverse types of tumors including breast cancer, suggesting that this enzyme plays a crucial role in cancer metabolism [64]. ACLY has been reported to have a strong expression in breast cancer tissue, with respect to adjacent normal tissues, and silencing ACLY expression in MCF-7 cell line suppressed cell viability and increased cell apoptosis [65]. Accordingly, a study reported that genetic or chemical inhibition of ACLY reduces, both in vitro and in vivo, proliferation, and tumor growth [66].
In recent work, Lucenay et al. demonstrated that cyclin E, an independent predictor of survival in patients with invasive breast cancer, upregulating ACLY activity leads to lipid droplet accumulation, a process positively correlated with tumor growth and development [67]. ACLY mRNA has been reported to be mostly expressed in the HER2-enriched subtype with respect to TNBC, linking the expression of this enzyme to the EMT process [65][66].
Several studies have highlighted the association between ACC 1/2 and FASN expression and activity with invasion, proliferation, and EMT [66][67][68][69][70]. Enhanced of both expression and activity of FASN are considered early events in breast cancer progression [71] and blocking FASN can induce antitumor effects in TNBC [68]. Additionally, inhibition of FASN by cerulenin can affect EMT [72] and reverse the hyperglycemia-induced EMT phenotype [73]. Fasnall, a selective FASN inhibitor, reduced the proliferation of breast cancer cells and modulated the lipidomic profile of these cells by increasing ceramide levels due to malonyl-CoA accumulation and consequent CPT-1 inhibition [74]. More recently, CRISPR/Cas9 knockout of FASN in MCF-7 cells demonstrated that FASN inhibition has a role in reducing proliferation, cell survival, cell size, cell cycle, migration, cell adhesion, and DNA replication [75].
A study conducted by Alò and collaborators demonstrated that FASN overexpression is associated with the stage of progression of breast cancer and that FASN expression can be used as a prognostic indicator for disease-free survival and overall disease survival [76]. In breast cancer stem cell sub-populations, high expression levels of ACC 1/2 and FASN have been correlated with increased cell survival and, in turn, with the formation of pre-malignant lesions [77]. Moreover, a decreased level of palmitic acid, associated with ACC1 and FASN gene silencing, can induce apoptosis in human breast cancer cells [70]. Interestingly, it has been reported that breast cancer susceptibility gene 1 (BRCA1) can exert its tumor suppressor function by preventing p-ACC1 dephosphorylation and, in turn, decreasing DNL [78].
ACC 1/2 and FASN expression in breast cancer cells is regulated by diverse growth factors and sex hormones through their corresponding receptors such as PR, ER, androgen receptor, and HER [79]. Based on this responsiveness, lipogenic enzyme expression is associated with molecular subtypes, and then with the malignant phenotype of breast cancers. Data suggest that in breast cancer cell lines overexpressing HER2, both FASN and ACC1 levels increased compared with cells in which HER2 expression is relatively low (such as MDA-MB-231) [80]. Indeed, induction of HER2 in MDA-MB-231 cells stimulates ACC1 expression via the PI3K/Akt pathway [81]. FASN upregulation in HER2-positive cells occurs throughout an SREBP-1-mediated mechanism. More recently, HER2 has also been shown to directly phosphorylate and activate FASN activity [82].
It must be pointed out that a metabolic transition that suppresses lipogenesis and promotes energy production is an essential component of metastasis in breast cancer. Indeed, Snail, a key inducer of EMT, has been related to ACC2 suppression and increased oxidation of mitochondrial fatty acids [83] and TGFβ1, which induces EMT, suppresses ACC in MCF-7 cells [84]. Furthermore, epithelial breast cancer cells with high expression of E-cadherin showed high expression of FASN, while mesenchymal cells with high expression of vimentin showed high expression of carnitine palmitoyltransferase-1 and therefore of β-oxidation [84]. A recent work reports that in both human and murine breast cancers, ACC1 inhibition, by increasing the level of acetyl-CoA, can favor acetylation and activation of the transcription factor Smad2, and thus EMT and metastasis [85].
It has been suggested that SCD1 may play a key role in the generation of the malignant phenotype as well as in the subsequent proliferation and survival of cancer cells [86]. Accordingly, SCD1 expression is enhanced in breast cancer tissues in situ compared to normal tissue [87][88] and SCD1 expression was associated with shorter survival times in breast cancer patients [89]. SCD1 was reported to be overexpressed in both HER2-enriched subtype [90][91] and in breast cancer cells that overexpress mucin-1 [92]. Inhibition of SCD1 activity or silencing its expression leads to anti-proliferation effects in breast cancer cell lines [93][94][95][96][97][98]. Moreover, ERα regulates SCD1 expression. Indeed, in vitro treatment of MCF-7 and T47D cell lines with 17β-estradiol induces SCD1 expression and modulates the cellular monounsaturated/saturated fatty acid ratio [99]. This was also observed in vivo, where the relative amounts of phosphatidylcholines (PC) (36:1) compared to PC (36:0) and that of PC (36:1) compared to lysoPC (18:0) were significantly higher in the cancerous areas characterized by higher levels of SCD1 expression compared to normal areas [100].
This entry is adapted from the peer-reviewed paper 10.3390/ijerph18073575
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30.
- Krug, K.; Jaehnig, E.J.; Satpathy, S.; Blumenberg, L.; Karpova, A.; Anurag, M.; Miles, G.; Mertins, P.; Geffen, Y.; Tang, L.C.; et al. Proteogenomic Landscape of Breast Cancer Tumorigenesis and Targeted Therapy. Cell 2020, 183, 1436–1456.e31.
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62.
- Johansson, H.J.; Socciarelli, F.; Vacanti, N.M.; Haugen, M.H.; Zhu, Y.; Siavelis, I.; Fernandez-Woodbridge, A.; Aure, M.R.; Sennblad, B.; Vesterlund, M.; et al. Breast cancer quantitative proteome and proteogenomic landscape. Nat. Commun. 2019, 10, 1600.
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70.
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412–424.
- Haukaas, T.H.; Euceda, L.R.; Giskeødegård, G.F.; Lamichhane, S.; Krohn, M.; Jernström, S.; Aure, M.R.; Lingjærde, O.C.; Schlichting, E.; Garred, Ø.; et al. Metabolic clusters of breast cancer in relation to gene- and protein expression subtypes. Cancer Metab. 2016, 4, 12.
- Dolfi, S.C.; Chan, L.L.-Y.; Qiu, J.; Tedeschi, P.M.; Bertino, J.R.; Hirshfield, K.M.; Oltvai, Z.N.; Vazquez, A. The metabolic demands of cancer cells are coupled to their size and protein synthesis rates. Cancer Metab. 2013, 1, 20.
- Zhou, Z.; Ibekwe, E.; Chornenkyy, Y. Metabolic Alterations in Cancer Cells and the Emerging Role of Oncometabolites as Drivers of Neoplastic Change. Antioxidants 2018, 7, 16.
- Naik, A.; Decock, J. Lactate Metabolism and Immune Modulation in Breast Cancer: A Focused Review on Triple Negative Breast Tumors. Front. Oncol. 2020, 10, 2668.
- Feng, W.W.; Kurokawa, M. Lipid metabolic reprogramming as an emerging mechanism of resistance to kinase inhibitors in breast cancer. Cancer Drug Resist. 2020, 3, 1–17.
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189.
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95.
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200.
- Jones, S.F.; Infante, J.R. Molecular Pathways: Fatty Acid Synthase. Clin. Cancer Res. 2015. clincanres.0126.2015.
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Lipid metabolism in cancer cells under metabolic stress. Br. J. Cancer 2019, 120, 1090–1098.
- Giudetti, A.M.; De Domenico, S.; Ragusa, A.; Lunetti, P.; Gaballo, A.; Franck, J.; Simeone, P.; Nicolardi, G.; De Nuccio, F.; Santino, A.; et al. A specific lipid metabolic profile is associated with the epithelial mesenchymal transition program. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2019, 1864, 344–357.
- Chen, M.; Huang, J. The expanded role of fatty acid metabolism in cancer: New aspects and targets. Precis. Clin. Med. 2019, 2, 183–191.
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890.
- Stefania, D.D.; Vergara, D. The Many-Faced Program of Epithelial-Mesenchymal Transition: A System Biology-Based View. Front. Oncol. 2017, 7, 274.
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751.
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197.
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384.
- Lampa, M.; Arlt, H.; He, T.; Ospina, B.; Reeves, J.; Zhang, B.; Murtie, J.; Deng, G.; Barberis, C.; Hoffmann, D.; et al. Glutaminase is essential for the growth of triple-negative breast cancer cells with a deregulated glutamine metabolism pathway and its suppression synergizes with mTOR inhibition. PLoS ONE 2017, 12, e0185092.
- Kung, H.-N.; Marks, J.R.; Chi, J.-T. Glutamine synthetase is a genetic determinant of cell type-specific glutamine independence in breast epithelia. PLoS Genet. 2011, 7, e1002229.
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749.
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA Synthetase 2 Promotes Acetate Utilization and Maintains Cancer Cell Growth under Metabolic Stress. Cancer Cell 2015, 27, 57–71.
- Kamphorst, J.J.; Chung, M.K.; Fan, J.; Rabinowitz, J.D. Quantitative analysis of acetyl-CoA production in hypoxic cancer cells reveals substantial contribution from acetate. Cancer Metab. 2014, 2, 23.
- Shukla, S.K.; Gebregiworgis, T.; Purohit, V.; Chaika, N.V.; Gunda, V.; Radhakrishnan, P.; Mehla, K.; Pipinos, I.I.; Powers, R.; Yu, F.; et al. Erratum to: Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab. 2014, 2, 23.
- Yoshii, Y.; Furukawa, T.; Yoshii, H.; Mori, T.; Kiyono, Y.; Waki, A.; Kobayashi, M.; Tsujikawa, T.; Kudo, T.; Okazawa, H.; et al. Cytosolic acetyl-CoA synthetase affected tumor cell survival under hypoxia: The possible function in tumor acetyl-CoA/acetate metabolism. Cancer Sci. 2009, 100, 821–827.
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.-L.; et al. Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365.
- Mwaikambo, B.R.; Yang, C.; Chemtob, S.; Hardy, P. Hypoxia Up-regulates CD36 Expression and Function via Hypoxia-inducible Factor-1- and Phosphatidylinositol 3-Kinase-dependent Mechanisms*. J. Biol. Chem. 2009, 284, 26695–26707.
- Chabowski, A.; Górski, J.; Calles-Escandon, J.; Tandon, N.N.; Bonen, A. Hypoxia-induced fatty acid transporter translocation increases fatty acid transport and contributes to lipid accumulation in the heart. FEBS Lett. 2006, 580, 3617–3623.
- Lewis, C.A.; Brault, C.; Peck, B.; Bensaad, K.; Griffiths, B.; Mitter, R.; Chakravarty, P.; East, P.; Dankworth, B.; Alibhai, D.; et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 2015, 34, 5128–5140.
- Gharpure, K.M.; Pradeep, S.; Sans, M.; Rupaimoole, R.; Ivan, C.; Wu, S.Y.; Bayraktar, E.; Nagaraja, A.S.; Mangala, L.S.; Zhang, X.; et al. FABP4 as a key determinant of metastatic potential of ovarian cancer. Nat. Commun. 2018, 9, 2923.
- Casciano, J.C.; Perry, C.; Cohen-Nowak, A.J.; Miller, K.D.; Vande Voorde, J.; Zhang, Q.; Chalmers, S.; Sandison, M.E.; Liu, Q.; Hedley, A.; et al. MYC regulates fatty acid metabolism through a multigenic program in claudin-low triple negative breast cancer. Br. J. Cancer 2020, 122, 868–884.
- Sp, N.; Kang, D.Y.; Kim, D.H.; Park, J.H.; Lee, H.G.; Kim, H.J.; Darvin, P.; Park, Y.-M.; Yang, Y.M. Nobiletin Inhibits CD36-Dependent Tumor Angiogenesis, Migration, Invasion, and Sphere Formation Through the Cd36/Stat3/Nf-Κb Signaling Axis. Nutrients 2018, 10, 772.
- Hellerstein, M.K. De novo lipogenesis in humans: Metabolic and regulatory aspects. Eur. J. Clin. Nutr. 1999, 53, s53–s65.
- Abu-Elheiga, L.; Jayakumar, A.; Baldini, A.; Chirala, S.S.; Wakil, S.J. Human Acetyl-CoA Carboxylase: Characterization, Molecular Cloning, and Evidence for Two Isoforms. Proc. Natl. Acad. Sci. USA 1995, 92, 4011–4015.
- Abu-Elheiga, L.; Almarza-Ortega, D.B.; Baldini, A.; Wakil, S.J. Human Acetyl-CoA Carboxylase 2: MOLECULAR CLONING, CHARACTERIZATION, CHROMOSOMAL MAPPING, AND EVIDENCE FOR TWO ISOFORMS. J. Biol. Chem. 1997, 272, 10669–10677.
- Abu-Elheiga, L.; Brinkley, W.R.; Zhong, L.; Chirala, S.S.; Woldegiorgis, G.; Wakil, S.J. The subcellular localization of acetyl-CoA carboxylase 2. Proc. Natl. Acad. Sci. USA 2000, 97, 1444–1449.
- Ntambi, J.M.; Miyazaki, M. Recent insights into stearoyl-CoA desaturase-1. Curr. Opin. Lipidol. 2003, 14, 255–261.
- Angelucci, C.; D’Alessio, A.; Iacopino, F.; Proietti, G.; Di Leone, A.; Masetti, R.; Sica, G. Pivotal role of human stearoyl-CoA desaturases (SCD1 and 5) in breast cancer progression: Oleic acid-based effect of SCD1 on cell migration and a novel pro-cell survival role for SCD5. Oncotarget 2018, 9, 24364.
- Shimomura, I.; Bashmakov, Y.; Ikemoto, S.; Horton, J.D.; Brown, M.S.; Goldstein, J.L. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc. Natl. Acad. Sci. USA 1999, 96, 13656–13661.
- Damiano, F.; Gnoni, G.V.; Siculella, L. Citrate carrier promoter is target of peroxisome proliferator-activated receptor alpha and gamma in hepatocytes and adipocytes. Int. J. Biochem. Cell Biol. 2012, 44, 659–668.
- Cheng, C.; Geng, F.; Cheng, X.; Guo, D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 2018, 38, 27.
- Guo, D.; Bell, E.H.; Mischel, P.; Chakravarti, A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr. Pharm. Des. 2014, 20, 2619–2626.
- Griffin, M.J.; Wong, R.H.F.; Pandya, N.; Sul, H.S. Direct Interaction between USF and SREBP-1c Mediates Synergistic Activation of the Fatty-acid Synthase Promoter. J. Biol. Chem. 2007, 282, 5453–5467.
- Ishii, S.; IIzuka, K.; Miller, B.C.; Uyeda, K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 15597–15602.
- Uyeda, K.; Yamashita, H.; Kawaguchi, T. Carbohydrate responsive element-binding protein (ChREBP): A key regulator of glucose metabolism and fat storage. Biochem. Pharmacol. 2002, 63, 2075–2080.
- Dentin, R.; Girard, J.; Postic, C. Carbohydrate responsive element binding protein (ChREBP) and sterol regulatory element binding protein-1c (SREBP-1c): Two key regulators of glucose metabolism and lipid synthesis in liver. Biochimie 2005, 87, 81–86.
- Airley, R.E.; McHugh, P.; Evans, A.R.; Harris, B.; Winchester, L.; Buffa, F.M.; Al-Tameemi, W.; Leek, R.; Harris, A.L. Role of carbohydrate response element-binding protein (ChREBP) in generating an aerobic metabolic phenotype and in breast cancer progression. Br. J. Cancer 2014, 110, 715–723.
- Chawla, A.; Repa, J.J.; Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors and Lipid Physiology: Opening the X-Files. Science 2001, 294, 1866–1870.
- Willy, P.J.; Umesono, K.; Ong, E.S.; Evans, R.M.; Heyman, R.A.; Mangelsdorf, D.J. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev. 1995, 9, 1033–1045.
- Janowski, B.A.; Willy, P.J.; Devi, T.R.; Falck, J.R.; Mangelsdorf, D.J. An oxysterol signalling pathway mediated by the nuclear receptor LXRα. Nature 1996, 383, 728–731.
- Joseph, S.B.; McKilligin, E.; Pei, L.; Watson, M.A.; Collins, A.R.; Laffitte, B.A.; Chen, M.; Noh, G.; Goodman, J.; Hagger, G.N.; et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc. Natl. Acad. Sci. USA 2002, 99, 7604–7609.
- Sugden, M.C.; Zariwala, M.G.; Holness, M.J. PPARs and the orchestration of metabolic fuel selection. Pharmacol. Res. 2009, 60, 141–150.
- Wang, Y.-X. PPARs: Diverse regulators in energy metabolism and metabolic diseases. Cell Res. 2010, 20, 124–137.
- Chao, L.; Marcus-Samuels, B.; Mason, M.M.; Moitra, J.; Vinson, C.; Arioglu, E.; Gavrilova, O.; Reitman, M.L. Adipose tissue is required for the antidiabetic, but not for the hypolipidemic, effect of thiazolidinediones. J. Clin. Investig. 2000, 106, 1221–1228.
- Kubota, N.; Terauchi, Y.; Miki, H.; Tamemoto, H.; Yamauchi, T.; Komeda, K.; Satoh, S.; Nakano, R.; Ishii, C.; Sugiyama, T.; et al. PPAR gamma mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol. Cell 1999, 4, 597–609.
- Miles, P.D.; Barak, Y.; He, W.; Evans, R.M.; Olefsky, J.M. Improved insulin-sensitivity in mice heterozygous for PPAR-gamma deficiency. J. Clin. Investig. 2000, 105, 287–292.
- Phan, L.M.; Yeung, S.-C.J.; Lee, M.-H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1–19.
- Burak Ozkaya, A.; Ak, H.; Aydin, S.; Aydin, H.H. Targeting Mitochondrial Citrate Transport in Breast Cancer Cell Lines. Anticancer. Agents Med. Chem. 2015, 15, 374–381.
- Zaidi, N.; Swinnen, J.V.; Smans, K. ATP-Citrate Lyase: A Key Player in Cancer Metabolism. Cancer Res. 2012, 72, 3709–3714.
- Wang, D.; Yin, L.; Wei, J.; Yang, Z.; Jiang, G. ATP citrate lyase is increased in human breast cancer, depletion of which promotes apoptosis. Tumor Biol. 2017, 39.
- Hatzivassiliou, G.; Zhao, F.; Bauer, D.E.; Andreadis, C.; Shaw, A.N.; Dhanak, D.; Hingorani, S.R.; Tuveson, D.A.; Thompson, C.B. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005, 8, 311–321.
- Lucenay, K.S.; Doostan, I.; Karakas, C.; Bui, T.; Ding, Z.; Mills, G.B.; Hunt, K.K.; Keyomarsi, K. Cyclin E Associates with the Lipogenic Enzyme ATP-Citrate Lyase to Enable Malignant Growth of Breast Cancer Cells. Cancer Res. 2016, 76, 2406–2418.
- Wang, Z.; Jiang, Q.; Dong, C. Metabolic reprogramming in triple-negative breast cancer. Cancer Biol. Med. 2020, 17, 44–59.
- Pandey, P.R.; Xing, F.; Sharma, S.; Watabe, M.; Pai, S.K.; Iiizumi-Gairani, M.; Fukuda, K.; Hirota, S.; Mo, Y.-Y.; Watabe, K. Elevated lipogenesis in epithelial stem-like cell confers survival advantage in ductal carcinoma in situ of breast cancer. Oncogene 2013, 32, 5111–5122.
- Chajès, V.; Cambot, M.; Moreau, K.; Lenoir, G.M.; Joulin, V. Acetyl-CoA Carboxylase α Is Essential to Breast Cancer Cell Survival. Cancer Res. 2006, 66, 5287–5294.
- Milgraum, L.Z.; Witters, L.A.; Pasternack, G.R.; Kuhajda, F.P. Enzymes of the fatty acid synthesis pathway are highly expressed in in situ breast carcinoma. Clin. Cancer Res. 1997, 3, 2115–2120.
- Menendez, J.A.; Vellon, L.; Mehmi, I.; Oza, B.P.; Ropero, S.; Colomer, R.; Lupu, R. Inhibition of fatty acid synthase (FAS) suppresses HER2/neu (erbB-2) oncogene overexpression in cancer cells. Proc. Natl. Acad. Sci. USA 2004, 101, 10715–10720.
- Zielinska, H.A.; Holly, J.M.P.; Bahl, A.; Perks, C.M. Inhibition of FASN and ERα signalling during hyperglycaemia-induced matrix-specific EMT promotes breast cancer cell invasion via a caveolin-1-dependent mechanism. Cancer Lett. 2018, 419, 187–202.
- Alwarawrah, Y.; Hughes, P.; Loiselle, D.; Carlson, D.A.; Darr, D.B.; Jordan, J.L.; Xiong, J.; Hunter, L.M.; Dubois, L.G.; Thompson, J.W.; et al. Fasnall, a Selective FASN Inhibitor, Shows Potent Anti-tumor Activity in the MMTV-Neu Model of HER2(+) Breast Cancer. Cell Chem. Biol. 2016, 23, 678–688.
- Gonzalez-Salinas, F.; Rojo, R.; Martinez-Amador, C.; Herrera-Gamboa, J.; Trevino, V. Transcriptomic and cellular analyses of CRISPR/Cas9-mediated edition of FASN show inhibition of aggressive characteristics in breast cancer cells. Biochem. Biophys. Res. Commun. 2020, 529, 321–327.
- Alo, P.L.; Visca, P.; Marci, A.; Mangoni, A.; Botti, C.; Di Tondo, U. Expression of fatty acid synthase (FAS) as a predictor of recurrence in stage I breast carcinoma patients. Cancer 1996, 77, 474–482.
- Li, G.; Zhao, F.; Cui, Y. Proteomics Using Mammospheres as a Model System to Identify Proteins Deregulated in Breast Cancer Stem Cells. Curr. Mol. Med. 2013, 13, 459–463.
- Brunet, J.; Vazquez-Martin, A.; Colomer, R.; Graña-Suarez, B.; Martin-Castillo, B.; Menendez, J.A. BRCA1 and acetyl-CoA carboxylase: The metabolic syndrome of breast cancer. Mol. Carcinog. 2008, 47, 157–163.
- Rochefort, H.; Chalbos, D. The Role of Sex Steroid Receptors on Lipogenesis in Breast and Prostate Carcinogenesis: A Viewpoint. Horm. Cancer 2010, 1, 63–70.
- Yoon, S.; Lee, M.-Y.; Park, S.W.; Moon, J.-S.; Koh, Y.-K.; Ahn, Y.-H.; Park, B.-W.; Kim, K.-S. Up-regulation of Acetyl-CoA Carboxylase α and Fatty Acid Synthase by Human Epidermal Growth Factor Receptor 2 at the Translational Level in Breast Cancer Cells. J. Biol. Chem. 2007, 282, 26122–26131.
- Corominas-Faja, B.; Cuyàs, E.; Gumuzio, J.; Bosch-Barrera, J.; Leis, O.; Martin, Á.G.; Menendez, J.A. Chemical inhibition of acetyl-CoA carboxylase suppresses self-renewal growth of cancer stem cells. Oncotarget 2014, 5, 8306–8316.
- Jin, Q.; Yuan, L.X.; Boulbes, D.; Baek, J.M.; Wang, Y.N.; Gomez-Cabello, D.; Hawke, D.H.; Yeung, S.C.; Lee, M.H.; Hortobagyi, G.N.; et al. Fatty acid synthase phosphorylation: A novel therapeutic target in HER2-overexpressing breast cancer cells. Breast Cancer Res. 2010, 12, R96.
- Yang, J.H.; Kim, N.H.; Yun, J.S.; Cho, E.S.; Cha, Y.H.; Cho, S.B.; Lee, S.-H.; Cha, S.Y.; Kim, S.-Y.; Choi, J.; et al. Snail augments fatty acid oxidation by suppression of mitochondrial ACC2 during cancer progression. Life Sci. Alliance 2020, 3, e202000683.
- Liu, Q.; Huo, H.; Ao, S.; Liu, T.; Yang, L.; Fei, Z.; Zhang, Z.; Ding, L.; Cui, Q.; Lin, J.; et al. TGF-β1-induced epithelial-mesenchymal transition increases fatty acid oxidation and OXPHOS activity via the p-AMPK pathway in breast cancer cells. Oncol Rep. 2020, 44, 1206–1215.
- Rios Garcia, M.; Steinbauer, B.; Srivastava, K.; Singhal, M.; Mattijssen, F.; Maida, A.; Christian, S.; Hess-Stumpp, H.; Augustin, H.G.; Müller-Decker, K.; et al. Acetyl-CoA Carboxylase 1-Dependent Protein Acetylation Controls Breast Cancer Metastasis and Recurrence. Cell Metab. 2017, 26, 842–855.e5.
- Igal, R.A. Roles of StearoylCoA Desaturase-1 in the Regulation of Cancer Cell Growth, Survival and Tumorigenesis. Cancers 2011, 3, 2462–2477.
- Mauvoisin, D.; Charfi, C.; Lounis, A.M.; Rassart, E.; Mounier, C. Decreasing stearoyl-CoA desaturase-1 expression inhibits β-catenin signaling in breast cancer cells. Cancer Sci. 2013, 104, 36–42.
- Luyimbazi, D.; Akcakanat, A.; McAuliffe, P.F.; Zhang, L.; Singh, G.; Gonzalez-Angulo, A.M.; Chen, H.; Do, K.-A.; Zheng, Y.; Hung, M.-C.; et al. Rapamycin regulates stearoyl CoA desaturase 1 expression in breast cancer. Mol. Cancer Ther. 2010, 9, 2770–2784.
- Holder, A.M.; Gonzalez-Angulo, A.M.; Chen, H.; Akcakanat, A.; Do, K.-A.; Fraser Symmans, W.; Pusztai, L.; Hortobagyi, G.N.; Mills, G.B.; Meric-Bernstam, F. High stearoyl-CoA desaturase 1 expression is associated with shorter survival in breast cancer patients. Breast Cancer Res. Treat. 2013, 137, 319–327.
- Monaco, M.E. Fatty acid metabolism in breast cancer subtypes. Oncotarget 2017, 8, 29487–29500.
- Kumar-Sinha, C.; Ignatoski, K.W.; Lippman, M.E.; Ethier, S.P.; Chinnaiyan, A.M. Transcriptome Analysis of HER2 Reveals a Molecular Connection to Fatty Acid Synthesis. Cancer Res. 2003, 63, 132–139.
- Pitroda, S.P.; Khodarev, N.N.; Beckett, M.A.; Kufe, D.W.; Weichselbaum, R.R. MUC1-induced alterations in a lipid metabolic gene network predict response of human breast cancers to tamoxifen treatment. Proc. Natl. Acad. Sci. USA 2009, 106, 5837–5841.
- Mason, P.; Liang, B.; Li, L.; Fremgen, T.; Murphy, E.; Quinn, A.; Madden, S.L.; Biemann, H.-P.; Wang, B.; Cohen, A.; et al. SCD1 inhibition causes cancer cell death by depleting mono-unsaturated fatty acids. PLoS ONE 2012, 7, e33823.
- Minville-Walz, M.; Pierre, A.-S.; Pichon, L.; Bellenger, S.; Fèvre, C.; Bellenger, J.; Tessier, C.; Narce, M.; Rialland, M. Inhibition of stearoyl-CoA desaturase 1 expression induces CHOP-dependent cell death in human cancer cells. PLoS ONE 2010, 5, e14363.
- Hess, D.; Chisholm, J.W.; Igal, R.A. Inhibition of stearoylCoA desaturase activity blocks cell cycle progression and induces programmed cell death in lung cancer cells. PLoS ONE 2010, 5, e11394.
- Fritz, V.; Benfodda, Z.; Rodier, G.; Henriquet, C.; Iborra, F.; Avancès, C.; Allory, Y.; de la Taille, A.; Culine, S.; Blancou, H.; et al. Abrogation of de novo lipogenesis by stearoyl-CoA desaturase 1 inhibition interferes with oncogenic signaling and blocks prostate cancer progression in mice. Mol. Cancer Ther. 2010, 9, 1740–1754.
- Scaglia, N.; Igal Ariel, R. Inhibition of Stearoyl-CoA Desaturase 1 expression in human lung adenocarcinoma cells impairs tumorigenesis. Int. J. Oncol. 2008, 33, 839–850.
- Zhao, J.; Zhi, Z.; Wang, C.; Xing, H.; Song, G.; Yu, X.; Zhu, Y.; Wang, X.; Zhang, X.; Di, Y. Exogenous lipids promote the growth of breast cancer cells via CD36. Oncol. Rep. 2017, 38, 2105–2115.
- Belkaid, A.; Duguay, S.R.; Ouellette, R.J.; Surette, M.E. 17β-estradiol induces stearoyl-CoA desaturase-1 expression in estrogen receptor-positive breast cancer cells. BMC Cancer 2015, 15, 440.
- Ide, Y.; Waki, M.; Hayasaka, T.; Nishio, T.; Morita, Y.; Tanaka, H.; Sasaki, T.; Koizumi, K.; Matsunuma, R.; Hosokawa, Y.; et al. Human breast cancer tissues contain abundant phosphatidylcholine(36:1) with high stearoyl-CoA desaturase-1 expression. PLoS ONE 2013, 8, e61204.