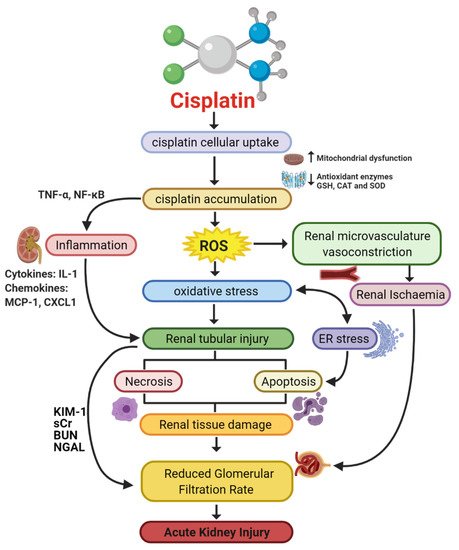
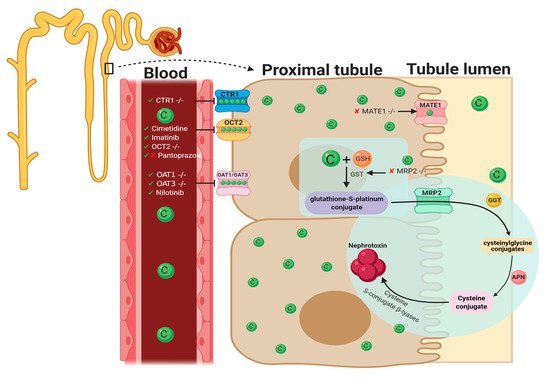
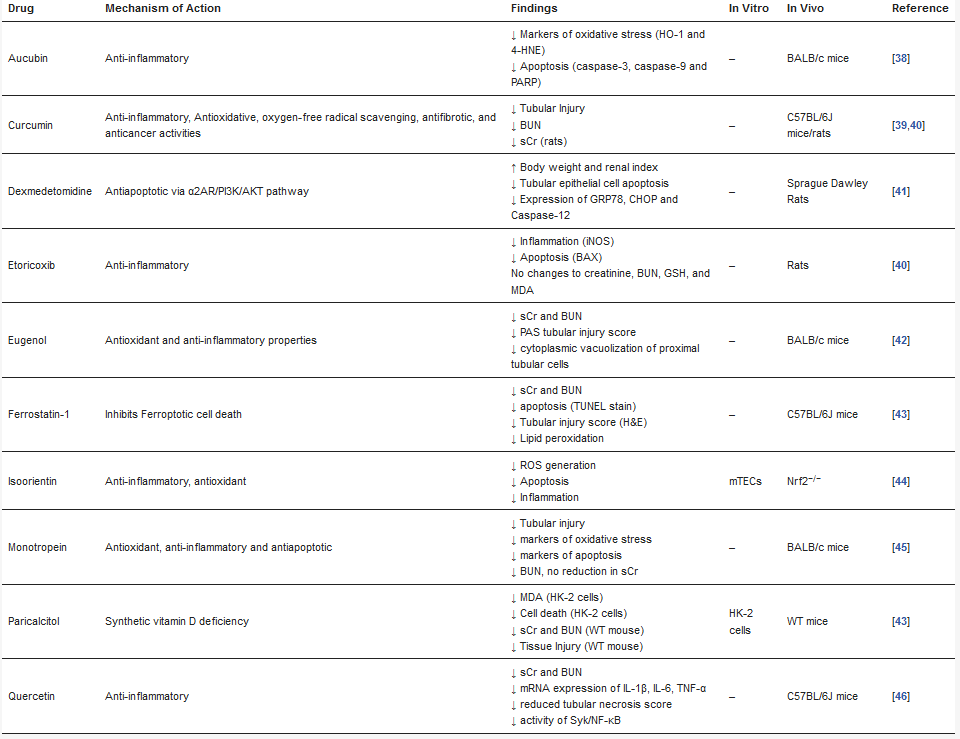
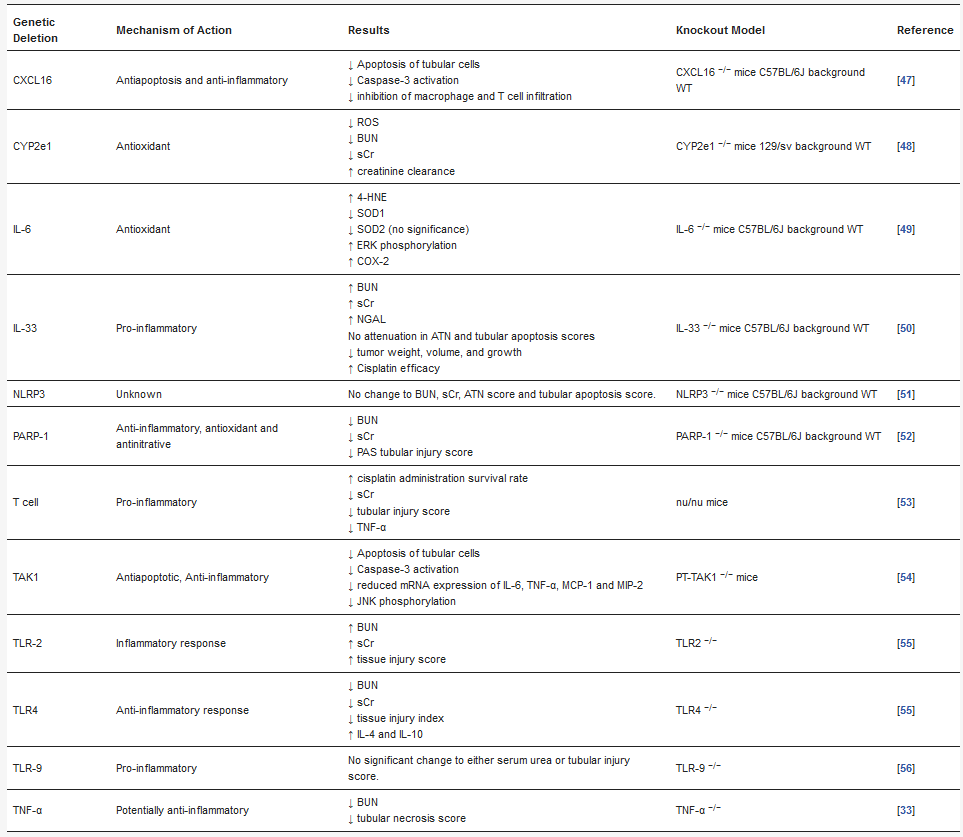
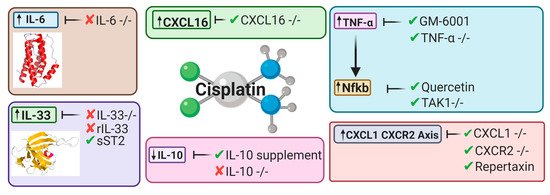
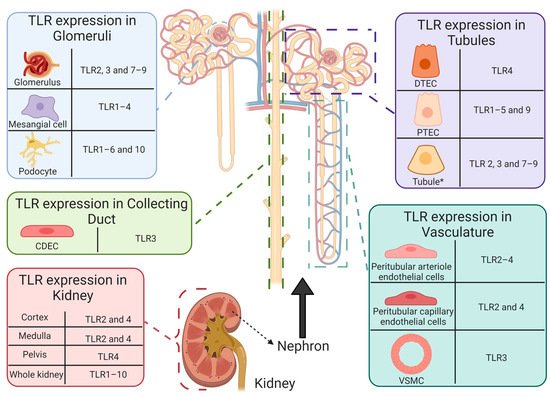
Since its discovery in 1997 [
238], TLR4 has been extensively investigated in the literature, and its structure, function, and signal transduction remain the most characterised and established of the TLRs [
239]. TLR4 plays an essential role in host Gram-negative immunity by identifying lipopolysaccharides, a lipid and polysaccharide conjugate that is a major component in the outer membrane of Gram-negative bacteria [
240]. However, the ability of TLR4 to recognize a repertoire of pathogens has been reported, including enveloped viruses [
241] and viral proteins (viral fusion proteins [
242] and glycoproteins [
243]), Gram-positive bacteria [
244], and helminths [
245]. Furthermore, TLR4 can respond to a broad range of DAMPs, including endoplasmin [
239], high-mobility group-1 [
246], and heat-shock protein 70 [
247], which have been shown to be upregulated during cisplatin treatment. Interestingly, TLR4 has also been shown to participate in transition metal sensing [
248], and metals, including nickel, cobalt, and platinum [
223,
248] have been observed as TLR4-activating ligands. Due to the increase in circulating DAMPs and cisplatin (a platinum-based chemotherapy) acting as TLR4 ligands, TLR4 has been implemented as a major contributor in driving pathogenesis and development of CIAKI through upregulation of inflammation and proinflammatory and subsequent renal dysfunction, renal tissue injury, and nephrotoxicity [
239]. A study involving TLR4-deficient mice administered a toxic dose of cisplatin (20 mg/kg) to induce acute renal failure within 72 h and reported significantly reduced markers of inflammation, nephrotoxicity, and renal function and decreased renal injury and histological abnormalities [
239]. To determine the effect that TLR4 deficiency has on cisplatin-induced renal dysfunction and structural changes, BUN and sCr were used as indicators of function [
239]. Severe renal failure (indicated by elevated levels of BUN and sCr) was observed between 48 and 72 h in WT after bolus dose of cisplatin and was accompanied by histological abnormalities including advanced tubular injury, cast formation, absence of brush-border membranes, shedding of tubular epithelial cells, necrosis of renal tubule cells, and dilation in renal tubules [
239]. Mice with TLR4 deficiency has significantly preserved renal function 72 h after administration to cisplatin, as shown by reduced BUN ad sCr concentrations and minimal histological changes. Thus, indicating that TLR4 contributes to structural and functional consequences during CIAKI [
239]. Furthermore, immunopathological consequences, including reduced leukocyte infiltration, decreased concentrations of cytokine and chemokine in serum (i.e., TNF-
α; IL-1
β; IL-2; IL-6; and IL-10), kidney (i.e., TNF-
α; IL-6; CCL5; MCP-10; and KC) urine (i.e., TNF-
α; IL-2; IL-6; CCL5; MCP-1; KC; and IP-10), and reduced activity of p38 MAPK and JNK phosphorylation was also observed in TLR4-deficient mice when compared to WT [
239]. Thus, suggesting that the potent inflammatory response initiated by TLR4 may be responsible for initiating CIAKI [
239]. Therefore, a tailored therapy encompassing a combination of cisplatin and a TLR4 inhibitor is an appealing approach to preserve renal structural integrity and preservation of function in CIAKI.
A potential TLR4 inhibitor to be used in conjunction with cisplatin, which has shown promising results in septic-induced AKI, is resatorvid (TAK242) [
249,
250]. TAK242 is a cyclohexene derivative [
251], which exerts its inhibitory effect by binding to the intracellular domain of TLR4. Upon binding, TAK242 causes a confirmation change in the cytoplasmic tail, which results in the inability of the bridging adaptor molecules TIR-containing adapter protein/myeloid differentiation factor-88 and translocating chain-associated membrane protein/TRIF to associate [
252], thus preventing TLR4 signal transduction and subsequent production and release of proinflammatory mediators [
252]. Administration of TAK242 to ovine models of Gram-negative bacteria resulted in enhanced renal function, demonstrated by abolishment of impaired Cr clearance in the urine, reduced BUN and sCr, prevention of renal hypoperfusion, and reduced swelling of endothelial cells in glomerular capillaries [
249,
250]. Additionally, a recent article has shown that TAK242 is able to enhance the cytotoxic effect of cisplatin in breast and ovarian cancer cells, while preventing its toxic effects of cells [
253]. Thus, suggesting that dual treatment with TAK242 and cisplatin could enable a reduced dose of cisplatin given to patients. Taken together, TAK242 should be further investigated in CIAKI, as it represents a pharmaceutical that could (i) prevent detrimental chronic inflammation, (ii) retain structural integrity and renal function, and (c) intensify the effect of cisplatin in CIAKI.