Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Medicine, General & Internal
Peripheral artery disease (PAD) is characterized by the development of atherosclerotic plaques in the lower-body conduit arteries. PAD is commonly accompanied by microvascular disease, which may result in poor wound healing, plantar ulcer development, and subsequent limb amputation.
- atherosclerosis
- endothelial dysfunction
- microcirculation
- peripheral vascular disease
- microcirculatory dysfunction
- microvascular disease
- critical limb ischemia
1. Introduction
Peripheral artery disease (PAD), which affects an estimated 200 million people globally [1], is characterized by the development of atherosclerotic plaques in the peripheral conduit arteries [2,3,4]. The most common manifestation of PAD is claudication, which includes symptoms such as leg pain, cramping, fatigue, pressure, and muscle weakness. These symptoms are often brought on by physical activity and are attenuated by rest [5]. In late-stage PAD, critical limb ischemia may develop, which occurs when tissue perfusion can no longer match the metabolic demand of the tissues below an arterial occlusion at rest. This often results in chronic ischemic pain, ulcers, and ultimately amputation [6]. It has been reported that those with PAD are at an increased risk for developing cerebral artery disease and coronary artery disease [7]. Additionally, patients with PAD are prone to increased rates of depression, which is strongly associated with all-cause mortality [8]. PAD is also independently associated with a 13.9-fold increase in amputation risk, which is further increased if PAD is confounded by microvascular disease [9].
The manifestation of microvascular disease is independent of the development of atherosclerosis in conduit arteries, but microvascular disease has been identified as a common phenomenon in those with PAD [9]. It has been documented that many adaptations occur in cutaneous microcirculation throughout the progression of PAD, including reduced capillary density in late-stage PAD [10]. The presence of microvascular dysfunction/disease in PAD is associated with poor wound healing and ischemic plantar ulcers [9]. The development of ischemic plantar ulcers may be dangerous for patients with PAD because of the potential for subsequent lower limb amputation. Therefore, understanding the mechanisms underlying microvascular dysfunction/disease and resultant ulcers in PAD may be important for the development of better treatments and therapeutics targeted at reducing symptoms and improving quality of life.
2. Skin Types and Microcirculation in PAD
Cutaneous microcirculation is responsible for perfusing dermal and epidermal cell structures and for full-body thermoregulation [11,16,17]. The majority of cutaneous microcirculation is in the upper horizontal plexus, which resides 1–2 mm deep to the epidermal surface [18]. The upper horizontal plexus is in charge of maintaining a large potential for temperature regulation [18,19]. Importantly, the ultrastructure of cutaneous microcirculation in the upper horizontal plexus is further complicated by whether the region is classified as glabrous or non-glabrous. This skin type classification and its subsequent microcirculatory properties may yield further insight regarding microcirculatory dysfunction and ulcer development in PAD.
Non-glabrous microcirculation: Non-glabrous skin, which contains hair follicles [11], is innervated by both a sympathetic vasoconstrictor and vasodilator system [20]. Several local factors contribute to the control of skin perfusion in non-glabrous skin, such as local temperature changes [21,22]. There is evidence indicating that non-glabrous microcirculation has limited capacity for autoregulation, given that perfusion pressure and skin blood flow changes exhibit a direct relationship [23]. However, non-glabrous skin does contain a unique regulatory feature, which is the venoarteriolar reflex (VAR) [20]. The VAR serves to combat blood pressure fluctuations, which may compensate for the deficiency in autoregulatory capabilities [20]. When orthostatic pressure is altered, specifically when cutaneous venule pressure increases, such as during a change in posture, the VAR, a local sympathetic reflex, is activated and causes vasoconstriction of arterioles, which protects capillary structures against the flux of blood brought about by the changing gravitational potential [24].
Glabrous microcirculation: Glabrous skin is non-hairy skin found on the plantar aspects of the feet, the palmar sides of the hands, and in various locations on the head [11]. Contrary to non-glabrous skin microcirculation, the glabrous microcirculation is thought to have autoregulatory capabilities but does not contain a VAR [20,25]. A structure entirely unique to glabrous skin is the arteriovenous anastomose (AVA) [26]. AVAs are direct conduits between dermal arterioles and venules, thus AVAs do not contribute to nutritive circulation like that of capillary networks and loops. AVAs contain thick concentric layers of smooth muscle and are densely innervated by sympathetic adrenergic nerves [26]. However, there is no active vasodilator system, so AVAs do not respond to metabolic vasodilators [27]. AVAs are thought to be under central control and are integral components of core temperature regulation [26]. In the thermoneutral temperature zone, the AVAs remain mostly closed, but during periods of extreme heat stress, the AVAs may become completely dilated (via release of sympathetic tone) to allow for increased blood volume closer to the external environment [26].
When the microcirculatory network becomes dysfunctional in chronic ischemia (i.e., PAD), the autoregulatory potential and function of VARs and AVAs may be attenuated. Midttun et al. (1997) examined the impacts of orthostatic pressure on glabrous skin microcirculatory characteristics in PAD [25]. Microcirculatory flow was assessed in the great toe at the level of the heart, above the heart, and below the heart. At the level of the heart, it was found that nutritive perfusion in claudicating patients was similar to healthy controls. When the great toe was lowered below the heart, the controls exhibited no change in cutaneous microcirculation perfusion, which was attributed to the absence of a VAR in the glabrous skin of the great toe. The maintenance of perfusion, despite orthostatic pressure changes in the great toe, was primarily attributed to autoregulation. However, claudicating patients with PAD demonstrated an increase in blood flow nearly 1.6x greater than what was measured at the level of the heart [25]. This lack of perfusion control in PAD may indicate attenuated autoregulatory potential in the glabrous microcirculation, considering that flow increased when the orthostatic pressure was increased.
The hydrostatic hypothesis, which is a proposed mechanism for plantar ulcer development in diabetics, states that ulcer development is caused by edema formation in the skin, due to increased hydrostatic pressures in the capillary beds [28]. While the effects of diabetes on microcirculation are not directly transferable to PAD, the loss of glabrous autoregulation in PAD may contribute to ulcer development in a similar manner as stated by the hydrostatic hypothesis. Without adequate autoregulation, changes in orthostatic pressure would lead to intermittently higher capillary pressures and hydrostatic pressures in the glabrous capillaries of the plantar foot in PAD [24]. It has also been stated that increased capillary hydrostatic pressures lead to an increase in basement membrane thickness [24]. An increase in basement membrane thickness would increase the diffusion distance for oxygen [29] and therefore could contribute to the progressive ischemia, tissue damage, and fibrosis associated with PAD.
Differentiating microcirculation between glabrous and non-glabrous skin will be crucial for understanding cutaneous perfusion and full-body thermoregulation [11,16,17], since glabrous and non-glabrous skin have unique control systems and structures. Studies investigating glabrous skin are useful for elucidating the mechanisms of plantar ulcer formation in PAD because the plantar foot is predominately glabrous [26]. Patients with PAD exhibit reduced capacity to autoregulate perfusion in the glabrous skin during changes in orthostatic pressure, which may be a key mechanism for plantar ulcer development in this population [25].
3. Microcirculation in PAD: Endothelial-Dependent and Endothelial-Independent Mechanisms
Patients with PAD exhibit reduced arterial pressures below an occlusion [30,31]. According to Poiseuille’s law, if resistance remains constant, a reduced pressure differential results in reduced flow [32]. However, it has been documented that patients with PAD (non-CLI) preserve both cutaneous microcirculation (flow) [24,25,28,33,34,35,36] and venous filling pressure [28] at rest (Table 1). Thus, to satisfy Poiseuille’s law, there must be a reduction in the resistance of the post-occlusion network to maintain flow [32] when the pressure differential is reduced [30,31]. This may indicate a compensatory mechanism in the vascular network below an occlusion in PAD. A change in arteriole tone, and therefore resistance, could influence microcirculation characteristics and thus it is important to elucidate any compensatory mechanism(s) that may affect microcirculation to better understand plantar ulcer development in PAD.
Table 1. Basal cutaneous capillary perfusion and pressure are preserved in non-critical limb ischemia (CLI) peripheral artery disease (PAD) patients compared to controls. Level of Disease is based on the Rutherford and Fontaine classification systems, which classify the progression of PAD into four major categories: Stage I is asymptomatic, Stage II is characterized by intermittent claudication, Stage III is characterized by rest pain, and Stage IV is characterized by tissue loss and ischemic ulcer development.
| Study | Measurement Tool | Measurement Location | Level of Disease | Outcomes |
|---|---|---|---|---|
| Cisek et al. [24] | Laser Doppler Flowmetry | Plantar aspect of great toe | Stage II | ↔ Basal perfusion |
| Rossi et al. [34] | Laser Doppler Flowmetry | Forearm and leg | Stage II | ↔ Basal perfusion |
| Husmann et al. [33] | Laser Doppler Flowmetry | Plantar aspect of great toe | Stage I–III | ↔ Basal perfusion |
| Rossi et al. [35] | Laser Doppler Flowmetry | Dorsum of the foot and forearm | Stage II | ↔ Basal perfusion |
| Midttun et al. [25] | Heat Washout and 133-Xenon Washout | Plantar aspect of great toe | Stage II | ↔ Basal perfusion |
| Graaff et al. [30] | Laser Doppler Flowmetry | Nail fold of hallux, ankle, toe | Stage II–IV | ↔ Capillary pressure |
| Urbanicic et al. [36] | Laser Doppler Flowmetry | Medial malleolus | N/A | ↔ Basal perfusion |
Microvascular response to post-occlusive hyperemia (reactive hyperemia): Post-occlusive hyperemia is a well-accepted assessment to non-invasively evaluate vascular endothelial function, which is one of the major vasodilation mechanisms. Rossi et al. (2005) revealed a potential compensatory mechanism while examining reactive hyperemia in patients with PAD [35]. A pneumatic cuff was applied to the upper thigh to induce acute ischemia of the peripheral tissue. Flow motion waveforms were decomposed into cardiac, respiratory, neurogenic, myogenic, and endothelial contributions [35]. This study reported that neurogenic, myogenic, and endothelial spectral powers were significantly higher in PAD compared to healthy subjects at rest. It was also reported that cardiac spectral power was significantly reduced in PAD compared to healthy subjects. There was no significant difference in respiratory contribution or basal microcirculation blood flux. Following post-occlusion hyperemia, neither group demonstrated a significant increase in endothelial, myogenic, or neurogenic activity, but there was a significant increase in cardiac activity in both PAD and healthy subjects. There was no difference in peak hyperemia between groups, but there was a significant difference in the time to peak hyperemia and the time to recovery after peak hyperemia between PAD and healthy subjects [35]. The previous study [35] suggests a compensatory mechanism in the post-occlusion network employed to maintain perfusion in microcirculation. The decrease in cardiac contribution to flow motion can be explained by the nature of an occlusion. If the pulse wave generated by the heart during systole were to represent a signal generated by the heart, an occlusion would distort the cardiac signal via changes in hemodynamics [35]. Thus, an occlusion would naturally downregulate the cardiac contribution to vasomotion. To compensate for the downregulation of cardiac contribution, the endothelium, smooth muscle, and nervous system activity are upregulated to maintain appropriate flow motion in the post-occlusion network. This compensatory mechanism is also supported by the findings regarding time to peak flux and recovery. Cardiac activity represents the highest-frequency contributor to flow motion [37], thus any reduction in cardiac contribution would decrease the rate at which flow motion could change. Rossi et al. (2005) reported an increased time to peak hyperemia and recovery after hyperemia in subjects with PAD [35], which likely indicates that low-frequency mechanisms of flow motion contribute more than high-frequency mechanisms of flow motion in PAD. Given that patients with PAD maintain normal flow characteristics in cutaneous microcirculation below an occlusion at rest [24,25,28,33,34,35,36], it can be inferred that the upregulation of endothelial, myogenic, and neurogenic activity [35] is adequate enough to maintain flow below an occlusion at rest, despite a decreased pressure differential [30,31].
Endothelial adaptations and the roles of hydrogen peroxide in vasodilation mechanisms: Endothelial-dependent vasodilation is positively coupled to blood pressure changes [36] and altered shear stress [38], so the increase in activation of endothelial cells observed by Rossi et al. (2005) [35] appears contradictory, considering that blood pressure and flow is reduced below an occlusion [30,31]. Although it is reported that capillary flow and filling pressure are preserved in PAD [24,25,28,33,34,35,36], flow would be reduced in all downstream blood vessels from the occlusion to the capillary beds. Since shear stress is directly coupled to vessel diameter and blood velocity, any vessels below an occlusion, with reduced flow, would experience a reduction in shear stress, which would result in less nitric oxide (NO), a potent endothelial produced vasodilator (Figure 1). Therefore, anywhere that a reduction in shear stress exists, there should be a decrease in the endothelial mediated NO contribution to vasodilation.
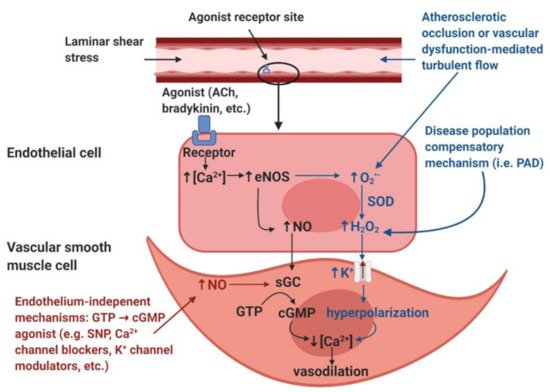
Figure 1. Illustration of the nitric oxide (NO)-mediated endothelial-mediated vasodilation pathway (black), the hypothesized disease population endothelial-dependent compensatory mechanism (blue), and endothelium-independent modulators of vasodilation (red). Image created with BioRender.com.
A possible explanation for the increase in endothelial-dependent vasodilation despite decreased shear stress and blood pressure may be explained by a “vasodilatory agonist switch,” which represents the shift of endothelial mediated vasodilation from nitric oxide to the endothelial derived hyperpolarization factor (EDHF), hydrogen peroxide (H2O2) [39] (Figure 1). It has been suggested that in states of disease, such as PAD, H2O2 may become favored over NO to facilitate vasodilation [39]. NO facilitates vasodilation by causing smooth muscle relaxation via calcium regulation, whereas H2O2 is capable of causing smooth muscle relaxation via hyperpolarization of the smooth muscle membrane [40]. Stress may be able to stimulate the production of H2O2 in endothelial cells [39]. This would provide an alternative route for the production of vasodilation agonists, independent of shear stress. It is likely that the tissues below an occlusion may be the main producers of inflammatory markers that increase the production of vasodilation agonists [1]. While it is supported that patients with PAD have elevated markers of inflammation [41], elevated H2O2 production is not directly coupled to PAD [41]. It may be that an agonist switch only occurs in highly progressed stages of PAD. Given that H2O2 is pro-inflammatory, a positive feedback loop of inflammation would likely result from H2O2 becoming the main agonist of vasodilation [39]. Whether inflammatory markers are capable of facilitating the production of NO is beyond the scope of this review.
NO-mediated vasodilation (black). The endothelial layer is stimulated by laminar shear stress and/or signaling molecules such as acetylcholine (ACh) and bradykinin. These stimuli induce an increase in intracellular calcium (Ca2+) within the endothelial cells. The increased [Ca2+] induces the activation of the calcium-dependent enzyme endothelial nitric oxide synthase (eNOS), resulting in the production of nitric oxide (NO). NO diffuses into the smooth muscle and binds to the receptor soluble guanylyl cyclase (sGC), which induces the accumulation of cyclic guanosine monophosphate (cGMP) from guanosine triphosphate (GTP). The accumulation of cGMP reduces intracellular [Ca2+] within the vascular smooth muscle cells and thus induces vasodilation.
Disease population compensatory mechanism (blue). In disease populations such as peripheral artery disease (PAD), it is hypothesized that the endothelial-derived hyperpolarization factor (EDHF) hydrogen peroxide (H2O2) may replace the NO-mediated vasodilatory pathway. Atherosclerotic occlusion or vascular dysfunction-mediated turbulent flow induces the formation of reactive oxygen species, specifically superoxide (O2•−). In addition, increased O2•− production is also thought to be partially mediated by eNOS uncoupling, a condition in which eNOS produces O2•− as opposed to NO. Dismutation by superoxide dismutase (SOD) or spontaneous dismutation occurs within the endothelial cell, in which O2•− and hydrogen ions produce H2O2. H2O2 diffuses into the vascular smooth muscle cell, which stimulates potassium (K+) channels, resulting in an efflux of K+ ions and resultant hyperpolarization of the vascular smooth muscle cells. This hyperpolarization results in a reduction in intracellular [Ca2+], which induces vasodilation.
Endothelium-independent modulators of vasodilation (red). Endothelium-independent mechanisms may also induce vascular smooth muscle relaxation. These pathways bypass the endothelium and include GTP → cGMP agonists such as sodium nitroprusside (SNP), Ca2+ channel blockers, K+ channel modulators, etc. The accumulation of cGMP from GTP will reduce intracellular [Ca2+] in the vascular smooth muscle cell, thus inducing vascular smooth muscle relaxation.
Vasodilation responses to heating: Local heating applications are another way to examine the reactivity of cutaneous microcirculation. Parshakov et al. (2016) examined the reactivity of PAD cutaneous microcirculation to local heating [3]. Temperature flux waveforms on the plantar surface of the great toe, isolating endothelial, myogenic, and neurogenic contributions to the local heating response were assessed [3]. Previous works have demonstrated a positive correlation between temperature waveforms and blood flow waveforms in the frequency domains related to endothelial, myogenic, and neurogenic activity [42]. Parshakov et al. reported that during heating, the relative contributions of the endothelium, smooth muscle, and nervous system were downregulated in PAD compared to healthy controls [3].
These findings initially seem contradictory to the work of Rossi et al. (2005), which demonstrated an upregulation of endothelial, myogenic, and neurogenic activity in PAD [35], but on further analysis, these findings are complementary. These findings can be explained by the operation limits of living organisms, which suggest that the variability of vasomotion has an upper and lower bound, between which it operates to maintain vascular homeostasis. If the mechanisms responsible for vasodilation reside at an elevated steady state [35], the maximum potential for vasodilation would be reduced because the system would continue to operate under the original physiological limits. If the potential to respond to a stimulus is attenuated, this would present as a downregulated response when a stimulus is imposed on the system. Therefore, the attenuated vascular reactivity to a heating stimulus observed by Parshakov et al. [3] seems to be explained by the elevated vasodilator steady state documented by Rossi et al. [35].
Receptor mediated vasodilation responses: Acetylcholine (Ach), a muscarinic receptor agonist, is considered to be an endothelial-dependent vasodilator, and sodium nitroprusside (SNP), an NO donor, is considered to be an endothelial-independent vasodilator (smooth muscle function) [40]. Thus, these two receptor agonists for endothelial-dependent and -independent vasodilation may elucidate the health of both the endothelium and smooth muscle. In two separate experiments, Rossi et al. (2002, 2005) examined maximal vasodilation in cutaneous microcirculation below an occlusion in patients with PAD via pharmacological-mediated vasodilation [34,35]. Through multistage vasodilation, it was found in both experiments that at mid to high stages, the vasodilatory response was significantly attenuated in PAD compared to healthy controls with both Ach and SNP administration [34,35]. The function representing the vasodilatory response to Ach administration resembled the upper end of a sigmoidal curve in the PAD group, indicating a limit, but increased without bound in the healthy controls. Limits were not clearly visible in the vasodilatory response to progressive SNP administration [34,35].
These results [34,35] further complement the notion of compensatory mechanisms existing below an occlusion in PAD. It was previously mentioned that patients with PAD exhibit a blunted response to local heating [3] and this was interpreted as a relationship between physiological limits and an elevated steady state [35], rather than damage to the vasodilatory mechanisms themselves. This reasoning can also be applied to the PAD vasodilatory response to the administration of Ach and SNP. In both experiments by Rossi et al. (2002, 2005), the PAD group achieved clear vasodilation limits, while control subjects did not demonstrate a vasodilatory limit [34,35]. Rather than attribute the observed limits [34,35] to endothelial dysfunction, this review suggests that these limits may indicate the interaction between an elevated resting vasodilation steady state and a constant physiological limit.
Blood flow and venous filling pressure are preserved in cutaneous microcirculation in patients with PAD [24,25,28,33,34,35,36]. In order to satisfy Poiseuille’s law, resistance below an occlusion must decrease to maintain flow in the wake of a decreased pressure differential [32]. A potential mechanism, which may lead to reduced resistance in the circulatory network below an occlusion, may be increased arteriole vasodilation. This review suggests that the increase in vasodilation steady state [35], limits to relative vasodilatory responses [34,35], and the preservation of cutaneous microcirculation blood flow [24,25,28,33,34,35,36] and venous filling pressure [28] are indicative of a compensation mechanism for which vasodilation is increased below an occlusion to decrease resistance and satisfy Poiseuille’s law [32]. In addition to this evidence, it has been observed that arterioles in patients with PAD, suffering from critical limb ischemia (CLI), are near maximally dilated at rest [43]. This may suggest a progressive compensation mechanism for which the vasodilation steady state below an occlusion is gradually upregulated in parallel to the progressive narrowing of the upstream occlusion, resulting in elevated dilation at rest. Developing an understanding of a compensatory mechanism, where basal arteriole tone in microcirculation is progressively shifted towards dilation, could be important for elucidating the progression of plantar foot ulcers in PAD (Figure 2).
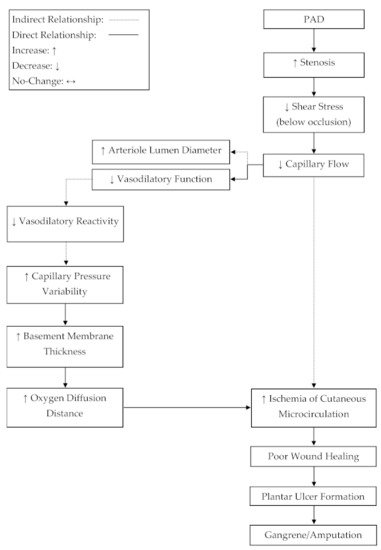
Figure 2. A proposed mechanism for progressive tissue ischemia below an occlusion in peripheral artery disease (PAD). The progressive narrowing of the conduit arteries and the reduced reactivity of arterioles, due to plaque development, may lead to ischemia-related, plantar ulcers and subsequent amputation.
This entry is adapted from the peer-reviewed paper 10.3390/ijerph18052384
This entry is offline, you can click here to edit this entry!