Human adenoviruses (hAds) are non-enveloped viruses with a diameter of 70 to 100 nm.
- oncolytic adenovirus
- cancer
- immunovirotherapy
1. Adenovirus (Ads) Vector Design
Human adenoviruses (hAds) are nonenveloped viruses with a diameter of 70 to 100 nm. The external protein shell of the virus is icosahedral, with 20 triangular faces, 30 edges, and 12 vertices, and this symmetry is made up in large parts by the major virus protein, hexon. hAds are members of the family Adenoviridae and are classified into the genus Mastadenovirus. There are 51 human Ad serotypes originally classified based on their ability to be neutralized by specific animal antisera. These can be further subdivided into seven species—or subgroups—(A to G), with a further subdivision of species B into subspecies B1 and B2 on the basis of their capacity to clump erythrocytes of humans, rats and monkeys as well as on the basis of their oncogenicity in rodents. More than 30 simian adenoviruses (sAds) display sequence identities to their human counterparts to such an extent that they have also been included in the taxonomy of human adenoviruses, within species B, C, E, and G [13]. hAds were initially isolated mainly from military forces with acute febrile respiratory disease and were subsequently associated with a number of clinical signs, including keratoconjunctivitis, gastroenteritis, hepatitis, meningoencephalitis, cystitis, upper and lower respiratory tract infections, and myocarditis, but also with noninflammatory conditions, such as obesity [14]. hAds infections are easily transmittable and, in some instances, highly contagious. Although the clinical courses are usually mild and self-limiting, infections may cause localized outbreaks with a critical course, occasionally leading to a lethal outcome even in the immunocompetent [15].
Ads are usually modified in specific regions [16], such as E1, E2A, E3, and E4 genes (Figure 1) [17]. E1, E2a, and E4 genes are essential for vector replications and are complemented in producer cell lines such as HEK-293 and their subsequently modified versions [17]; vectors with deletion of the above-mentioned genes are replication-defective but still maintain the ability to induce a strong host immune response towards both vectors and transgenes. The E3 gene is dispensable for vector replication and is deleted to increase vector capacity. Subsequent generations of Ad vectors have been developed leading to safer, less toxic, and more capable vectors. In first-generation adenoviral (FG-Ad) vectors, the E1 gene is deleted and replaced with the transgene; the packaging capacity of FG-Ad vectors is limited because only a limited amount of virus genome is deleted (8.2 kb) [18], and, consequently, the inserted transgene can be of limited size. Second-generation adenoviral (SG-Ad) vectors have deletions in E1, E3, and E2 or E4 genes, resulting in a reduced possibility of reversion to a replication-competent Ad and an increased room to accommodate larger transgenes (up to 10 kb). Finally, using helper-dependent adenoviral (HD-Ad) vectors, the whole genome can be substituted with DNA of interest. HD-Ad vectors contain only cis-acting Ads sequences necessary for viral DNA replication and a packaging and can accommodate up to 35 kb of foreign DNA [7,19]. Production of HD-Ad vectors requires a helper virus (HV) that provides all the protein products necessary for replication [20] expressing the Cre recombinase that eliminates the possibility of HV genome packaging. High accommodation capacity is one of the principal advantages of HD-Ads, together with the ability to efficiently transduce a wide variety of cell types, regardless of the cell cycle [21]. The complete absence of the viral genes attenuates cellular toxicity and host response, resulting in decreased virus clearance [22]; in addition, the minimal overlap with the adenoviral HV genome abrogates the possibility of generating wild-type (wt) Ad by recombination events.
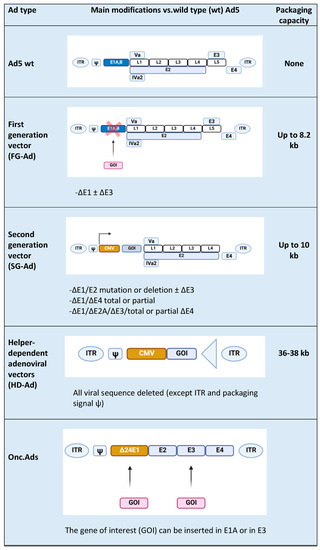
Figure 1. Types of Ad5-derived vectors and their characteristics: wild type (wt), first generation (FG-Ad), second generation (SG-Ad), and helper-dependent (HD-Ad) adenoviral vectors. At the end of the table is a schematic representation of an Onc.Ad. The figure was created with BioRender.com.
Although Ads have been originally developed for transgene delivery in gene therapy applications, they have become interesting candidates as an anticancer agent. In fact, the development of replicative Onc.Ads to selectively replicate in cancer cells has been one of the early innovative strategies for cancer gene therapy [23]. Expression of E1A, a protein with a pivotal role in the early virus replicative cycle, under the control of a promoter typically overexpressed in specific cancer cells and usually not expressed in the normal tissues was the first strategy for cancer targeting [1]. A subsequent strategy to develop tumor-selective Onc.Ads consisted in the expression of a defective E1A unable to support replication in normal cells but able to lead to replication in the absence of the retinoblastoma protein (pRb) typically lacking in cancer cells. E1A-defective production is obtained with a deletion of 24 bp on this gene (E1A-d24) and the addition of an E2F promoter before the E1A-d24 gene [1].
The main feature of Onc.Ads that leads to their antitumoral effects consists in the ability to induce an immune response against cancer cells in different ways, both modulating the tumor microenvironment (TME) and through the release of tumor-associated antigens (TAAs) and neoantigens that are subsequently processed principally by antigen-presenting cells (APCs). In fact, Onc.Ads induce different types of immunogenic cell death (ICD), such as necrosis, necroptosis, pyroptosis, autophagic cell death, and immunogenic apoptosis. Although Onc.Ads have a significant effect on tumor cells’ growth and tumor size in vivo, they are usually unable to eradicate the whole tumor. In order to potentiate Onc.Ads effects, a local co-administration with HD-Ad vectors expressing a variety of antitumoral proteins revealed to be a promising strategy [20]. Indeed, the synergistic effect of Onc.Ads/HD-Ad vectors administration allows the amplification and packaging of HD-Ad vectors in cancer cells and overcomes the limitations of each virus: the reduced capacity of Onc.Ads and the inability of HD-Ad vectors to replicate in tumors [20].
2. Exploring the Tumor Microenvironment and Its Modulation by Onc.Ads
Since Rudolf Virchow discovered the presence of leukocytes in neoplastic tissues and proposed the link between chronic inflammation and tumorigenesis [24], a comprehensive understanding of the TME of solid tumors has attracted researchers’ attention [25]. The TME is a heterogeneous cellular environment in which the tumor propagates. Solid malignant tumors include not only tumor cells but also several non-transformed cells, including mesenchymal cells (cancer stem cells (CSCs), mesenchymal stem cells (MSCs), endothelial cells (ECS), fibroblasts, and myofibrobasts) that contribute to tumor cells’ growth. Often, the TME contains innate and adaptive immune cells including dendritic cells (DCs), mast cells (MCs), macrophages, neutrophils, T-cells, B-cells, natural killer (NK) cells, and myeloid-derived suppressor cells (MDSCs). The TME also includes surrounding blood vessels, proteins of the extracellular matrix (ECM), and a number of signaling molecules including cytokines and chemokines. Cancer cells along with T regulatory cells (Tregs), MDSCs, adipocytes, and tumor-associated macrophages (TAM) can hinder immune control of tumors by producing and releasing cytokines, such as interleukin-10 (IL-10), chemokines, such as chemokine C-X-C motif ligand 12 (CXCL12), growth factors, such as transforming growth factor beta (TGF-β), matrix remodeling factors, such as collagen, fibronectin, and fibrin, and other soluble factors, such as adenosine, into the TME [26,27]. The final effect consists in a strong immunosuppressive identity in the last phase (elimination) of the cancer immunoediting process of the tumor niche. In this environment, the immune system fails to recognize TAAs and tumor-associated neoantigens because tumor cells have devised ways to escape immune surveillance. In particular, TGF- β and IL-10 mediate an anti-inflammatory response by dampening the activity of tumor suppressor cells, such as cytotoxic T lymphocytes (CTLs) and NK cells, and enhancing the activity of tumor-promoting cells such as Tregs and tumor-associated neutrophils (TANs) [28,29].
As previously mentioned, the main advantage of using oncolytic viruses (OVs) is their ability to modulate the TME rendering it less immunosuppressive [30]. OVs can preferentially infect and kill cancer cells as result of the inhibition of the dysfunctional Type I IFNs signaling [31]; however, their main ability consists in inducing a response from the immune system impaired by the hostile and highly immunosuppressive environment of the tumor milieu. In fact, after a successful tumor infection, an inflammatory reaction is triggered because OVs are able to induce a particular form of apoptosis better known as immunogenic cell death (ICD). During this process, the OV-mediated cancer cell lysis releases TAAs into the microenvironment allowing the immune system to recognize them and to generate a response, breaking down the immuno-editing process. Specifically, TAAs recruit and activate DCs with consequent stimulation of specific lymphocytes, evoking an effective anti-tumor response. Then, the ICD is not sterile, but it triggers the endoplasmic reticulum with the consequent release of some dangerous metabolites called damage-associated molecular patterns (DAMPs) such as calreticulin, ATP, and HMGB1 [32]. Furthermore, ICD mediated by OVs is associated with the release of pathogen-associated molecular patterns (PAMPs) that bind pattern recognition receptors (PRRs) on innate immune cells and function as danger and eat-me signals. The recognition of these key metabolites by the APCs in the tumor microenvironment contributes to trigger an immune response. Therefore, virus-mediated ICD leads to an inflammatory response and a localized cytokine production followed by infiltration of innate immune cells and CTLs that help to shape the TME in a less immunosuppressive manner [33]. Despite the multipower of OVs, all that glitters is not gold because the antitumor- immunity generated by OVs is hampered by the classical anti-viral response from normal cells. The activation of the immune system destroys infected cancer cells but also clears the OV, reducing the therapeutic efficacy [32]. This immunological system has to be manipulated in order to balance the anti-viral response with the anti-tumoral response. This can be obtained by designing OVs that can replicate and spread within tumors quickly to induce maximal anti-tumor effect before clearance [34] or by increasing the recruitment of immune cells, which will kill the infected cells (i.e., tumor cells), potentiating the direct lysis of neoplastic cells by viral infection itself. The latter can be improved by arming viruses with immunostimulatory cytokines, chemokines, or immune-activating ligands and bispecific T-cell engager (BiTE) molecules in order to catch more immune components to the tumor site. In the next sections, we discuss the progress made in arming oncolytic adenoviruses and the successful combinations with other immunotherapy solutions.
2.1. Armed Oncolytic Adenoviruses with Immunostimulatory Cytokines and Chemokines
The current scientific trend is to attempt an increase of the antitumoral immune response taking advantages of different agents that can counteract cancer immune escape. Although OVs are able to induce anti-cancer immunity by multiple mechanisms, as described in the previous section, recent updates of clinical trials involving OVs confirm their modest activity as a monotherapy. This can be explained by the inability to optimally infect cancer cells due to (i) neutralizing antibodies, (ii) other antiviral clearance mechanisms, (iii) physical barriers that prevents OVs to reach their entry receptors, or due to viral intrinsic factors such as (iv) engineered cancer-selectivity or transgene expression that can reduce viral fitness and (v) expression of potent transgene(s) that may result in a significant immune response with premature clearance of the OV [35]. In this section, we review the current design strategies to harness the potential of oncolytic adenoviruses for cancer immunotherapy. To effectively trigger the immune response necessary for the removal of tumor cells, it is necessary to not only trigger an immune response but also to recruit immune cells. With this aim, many OVs have been modified to express immunostimulatory transgenes, such as interleukins.
Using immunostimulatory cytokines has become an increasingly promising approach in cancer immunotherapy because they indirectly activate tumor-specific T-lymphocytes capable of rejecting tumor cells from patients with a low tumor burden or because they protect patients from a recurrence of the disease. One of the most promising cytokines for arming oncolytic viruses is granulocyte macrophage colony stimulating factor (GM-CSF) [33,36]. Its pro-inflammatory activity is primarily due its role as a growth and differentiation factor of myeloid lineage cells and the granulocyte and macrophage populations in particular [37]. The ability of GM-CSF to enhance antitumor immunity via a T-cell-mediated mechanism has been potentiated by its local expression by OV, improving DC migration and maturation and eventually improving priming of the T-cell response [38]. Various Ads have been successfully armed with GM-CSF, such as ONCOS-102, currently in a phase I trial in combination with pembrolizumab (NCT03003676). Interesting data in support of the ongoing clinical study mentioned were given by L. Kuryk et al., who observed a synergistic anti-tumor effect in the humanized mice treated with the combination of ONCOS102 and pembrolizumab, as demonstrated by reduced tumor volumes [39].
Originally characterized as a potent inducer of natural killer [40] cell cytotoxic activity, interleukin 12 (IL-12) has been used for arming OVs. IL-12 is now recognized as a key regulator of cell-mediated immune response and a bridge between innate and adaptive immunity [41]. Because of its role as major orchestrator of Th1-type immune response against cancer [40], IL-12 is an attractive protein candidate for cancer therapies [42]. Studies conducted in a rat model of thyroid cancer showed that delivery of IL-12 gene with adenovirus (AdIL-12) was efficacious to elicit systemic anti-tumor immunity, unlike treatment with AdGM-CSF with cells expressing IL-12 or GM–CSF, which elicited only local effects. Chemokines constitute the largest family of cytokines, with approximately 50 endogenous chemokine ligands in humans and mice [43]. These small secreted proteins mediate immune cell trafficking and lymphoid tissue development. Different immune cell subsets migrate into the tumor microenvironment via interaction between chemokines and chemokine receptors, and these populations regulate the tumor immune response in a spatiotemporal manner, thus affecting disease progression and therapeutic results [44]. Different chemokines, such as CCL5 and CCL19, have been expressed in various types of virus; in particular, CCL20 and CCL21 have shown to enhance anti-tumor effects when used to arm Onc.Ads [45,46]. The generation of effective anti-tumor immune responses is a complex process dependent upon the coordinated interaction of various subsets of effector cells. As such, CCL21 and IL-21 are potent activators of the immune system when used together for tumor therapy. Multigene-armed oncolytic adenoviruses are capable of efficiently generating a productive antitumor immune response. Li et al. armed an oncolytic adenovirus with the chemokine (C-C motif) ligand 21 (CCL21) and with Interleukin 21 (IL-21) that was able to induce oncolytic effects and a tumor-specific cytotoxic T-lymphocytes (CTLs) response in vitro [45]. A similar strategy that combined CCL20 and CD40L was adopted resulting in an enhanced growth suppression of TERT-positive tumor cells [46].
2.2. Arming OVs with Immune-Activating Ligands and Bispecific T-Cell Engager (BiTE) Molecules
OVs have been shown to exert beneficial immunologic responses, including induction of anti-tumor T-cells and modulation of the tumor microenvironment from Th2 to Th1, which has been suggested to contribute to breakage of tolerance in tumors [47,48,49]. Nevertheless, oncolysis per se is usually not enough for immunologic eradication of advanced tumors and its action could be increased by arming the virus with immune stimulatory molecules. One of the most investigated immune-activating ligands is CD40L because it constitutes an interesting target in cancer immunotherapy because of its ability to stimulate Th1 immunity via maturation of dendritic cells and to drive M2 to M1 macrophage differentiation [50]. CD40 is a member of the tumor-necrosis factor (TNF) receptor family and is expressed on APCs, such as DCs and myeloid cells [51]. APCs greatly increase their antigen-presentation and costimulatory capacity and allow for efficient CD8+ CTL priming by signaling through CD40 [52]. In addition, CD40L is expressed on activated CD4+ T-cells, B-cells, and NK-cells as well as memory CD8+ T-cells [51]. Many OVs and viral vectors armed with CD40L have been tested in clinical [53,54,55,56] and preclinical [50,57,58] settings and have been shown to exert multiple antitumoral activities including tumor growth control, cancer cell apoptosis, induction of T-cell responses, increase in T-effector/T-reg cell ratios, and upregulation of Th1 cytokines. For instance, Pesonen et al. treated nine patients with refractory solid tumors using an OV armed with CD40L (CGTG-401) intratumorally, reporting that 83% of patients showed some disease control and experienced some grade 1 to 2 adverse events. However, induction of a tumor-specific T-cell response was observed in the majority of patients [53]. Furthermore, NG-350A, an Onc.Ad expressing a full-length agonist anti-CD40 antibody at the site of virus replication, is under investigation in a phase I clinical trial (NCT03852511).
The tumor necrosis factor receptor superfamily, member 4 (TNFRSF4), also known as CD134 and OX40 receptor, is another member of the TNFR-superfamily of receptors that have gained interest as therapeutic target molecules for cancer immunotherapy. OX40 is not constitutively expressed on resting naïve T-cells and plays a key role in the survival and homeostasis of effector and memory T-cells, and it regulates the differentiation and function of Foxp3+ Tregs [59]. H. Jiang et al. have recently showed that OX40L-armed Onc.Ad (Delta-24-RGDOX) has a stronger anticancer efficacy compared to its predecessor Delta-24-RGD, triggering a greater tumor-specific lymphocyte activation and a proliferation of TAAs-specific CD8+ T-cells in two mouse glioma models [60]. In the same model, a synergistic therapeutic effect was observed by the intra-tumoral injection of Delta-24-RGDOX and an anti-PD-L1 antibody [61]. Therapeutic efficacy of Delta-24-RGDOX has been subsequently evaluated in subcutaneous and intracranial melanomas. Localized treatment of the subcutaneous melanoma inhibited growth of the intracranial ones, suggesting a strong systemic immunity in syngeneic glioma mouse models. Currently, a phase I trial is going on to evaluate the effects of Delta-24-RGDOX treatment in patients with recurrent glioblastoma (NCT03714334) and a phase II trial is evaluating the effects of pembrolizumab together with Ad5-DNX-2401 or Delta-24-RGD (NCT02798406) as reported in Table 1. Oncolytic virotherapy is being evaluated as a therapeutic approach in models for aggressive pediatric brain tumors, such as pediatric high-grade glioma (pHGG) and diffuse intrinsic pontine gliomas (DIPGs), with encouraging results in mouse models [62]. These data led to a phase I/II clinical trial for newly diagnosed diffuse intrinsic pontine gliomas (DIPG) (NCT03178032).
Table 1. Current clinical trials including oncolytic viruses (OVs) combined with immune checkpoint inhibitors. Clinical trials reported as completed are not listed.
| OV Type | Genetic Modification | Checkpoint Inhibitor | Indication | Clinical Phase | NCT Number |
|---|---|---|---|---|---|
| Herpes simplex virus 1 | Deletions in ICP34.5 and ICP47 and transgenic expression of GM-CSF | Pembrolizumab (anti-PD1) | Unresectable Stage IIIB–IV melanoma | III | NCT02263508 |
| T-VEC | Nivolumab (anti-PD1) | Lymphomas and some rare cutaneous tumors | II | NCT02978625 | |
| Pembrolizumab | Advanced melanoma progressed on anti-PD1/L1 based therapy | II | NCT02965716 | ||
| Pembrolizumab | Metastatic squamous cell carcinoma of the head and neck | I | NCT02626000 | ||
| Ipilimumab (anti-CTLA4) | Melanoma | I/II | NCT01740297 | ||
| Atezolizumab (anti-PDL1) | Breast cancer | I | NCT03802604 | ||
| Ipilimumab and nivolumab | Before surgery of localized breast cancer | I | NCT04185311 | ||
| Vaccinia virus Pexa-Vec |
TK deletion and expression of GM-CSF and β-galactosidase | Ipilimumab | Metastatic solid tumors | I | NCT02977156 |
| Durvalumab (anti-PD1)-Tremelimumab | CRC | I/II | NCT03206073 | ||
| (Anti-CTLA4) nivolumab | HCC | I/II | NCT03071094 | ||
| Cemiplimab (anti-PD1) | RCC | I | NCT03294083 | ||
| Vesicular stomatitis virus (VSV) | Engineered to express Na+/I− symporter (NIS) and human | Avelumab | Refractory solid tumors | I | NCT02923466 |
| Interferon Beta (VSV-IFNβ-NIS) | Pembrolizumab | Refractory NSCLC and HCC | I | NCT03647163 | |
| Reovirus reolysin | None | Nivolumab | Relapsed/refractory multiple myeloma | I | NCT03605719 |
| Adenovirus (Ad) ONCOS-102 | Onc.Ad expressing GM-CSF | Pembrolizumab | Advanced or unresectable melanoma | I | NCT03003676 |
| CG0070 | Onc.Ad with a tumor specific promoter expressing GM-CSF | Pembrolizumab | NMIBC | II | NCT04387461 |
| Ad-p53 | Ad. expressing p53 | Pembrolizumab | HNSCC | I/II | NCT02842125 |
| PD-1/PD-L1 Inhibitors | Lymphoma | II | NCT03544723 | ||
| Ad-MAGEA3 | Ad. expressing MAGE-A3 with MG1-MAGEA3 | Pembrolizumab | NSCLC | I/II | NCT02879760 |
| Pembrolizumab | Metastatic melanoma squamous cell skin carcinoma | I | NCT03773744 | ||
| Ad5-DNX-2401 or Delta-24-RGD | Ad. expressing an Integrin-binding RGD-4C motif | Pembrolizumab | GBM and GS | II | NCT02798406 |
CRC: colon rectal cancer; HCC: hepatocellular carcinoma; RCC: renal cell carcinoma; NSCLC: non-small cell lung cancer; NMIBC: non-muscle invasive bladder cancer; HNSCC: head and neck squamous cell carcinoma; MG1-MAGEA3: MG1 maraba oncolytic virus expressing melanoma-associated antigen 3 (MAGEA3); GBM: glioblastoma; GS: gliosarcoma.
CD40L has also been evaluated in combination with an additional co-stimulatory molecule, named 4-1BBL, to arm OVs [61]. 4-1BBL belongs to the TNFR family and is expressed on activated T-cells. Signaling through 4-1BB/4-1BBL stimulates T-cell expansion, acquisition of effector function, and survival [12]. The virus LOAd703, armed with CD40L and 4-1BBL, was shown to act as a potent immune activator in in vivo xenograft models of human pancreatic cancer. Such a double-armed virus efficiently reduced established tumors and could be combined with gemcitabine for additional effect. Currently, LOAd703 is undergoing two phase I/II clinical trials in patients with pancreatic cancer (NCT02705196) and in patients with pancreatic adenocarcinoma, ovarian cancer, biliary carcinoma, or colorectal cancer (NCT03225989). Finally, another costimulatory molecule that has successfully been used to arm OVs is glucocorticoid-induced tumor necrosis family receptor family-related gene (GITR) [63]. GITR is a modulator of immune response and inflammation; functional testing with specific antibodies showed that only an anti-GITR antibody could inhibit immune suppressive activity of an immunosuppressive T-cell population, T-regulatory cells (T-reg). This population, which expresses both CD4 and CD25, has been implicated in protecting tumors from immune attack and in supporting their growth in mouse models.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22052517