The efficacy and safety of vitamin K antagonists (VKAs) as oral anticoagulants (OACs) depend on the quality of anticoagulation control, as reflected by the mean time in therapeutic range (TTR).
- oral anticoagulants
- microbial metabolites
- vitamin k
1. Introduction
For many years, the only options available for oral anticoagulation (OAC) therapy were the vitamin K antagonists (VKAs), and they are still widely used for several conditions. The most common indication is stroke prevention in atrial fibrillation (AF) [1], so the majority of AF patients require long-term OAC, as suggested by guidelines [2][3]. Despite the introduction of direct-acting oral anticoagulants (DOACs), the use of VKAs in AF is common in some countries, for example in Eastern Europe and Southern Europe [4]. Aside from AF patients, there is also a wide range of patients for whom VKAs are the recommended OAC therapy.
Similarly, in patients with history of thrombosis and diagnosed with antiphospholipid syndrome (APS), the use of rivaroxaban was associated with an increased risk of recurrent thrombotic events, compared with warfarin [5]. In the absence of evidence for apixaban, edoxaban, and dabigatran, all DOACs are not recommended in patients with APS, particularly in high-risk patients (those who present a positive test for all 3 antiphospholipid tests: lupus anticoagulant, anticardiolipin antibodies, and anti-beta 2 glycoprotein I antibodies). In addition, APS patients taking a DOAC should be carefully evaluated and the possibility of switching to a VKA should be considered. Thus, VKA therapy remains the first-line treatment for a first or recurrent APS-related venous thrombotic event [6]. Finally, DOACs are currently more expensive than treatment with VKA and in the context of venous thromboembolism (VTE), there is no reimbursement for DOACs in several countries [7][8]. Consequently, an important proportion of VTE patients who could be potential users of DOACs are actually taking VKAs.
In patients who are prescribed VKAs, the quality of anticoagulation control is central to prevent bleeding and thromboembolic events [9]. There is evidence for gut microbiota influencing the progression or development of some cardiovascular diseases [10][11], but there are limited data linking gut microbiota to the quality of anticoagulation in VKA users.
2. Effect of Metabolites Produced by Gut Microbiota on VKAs Drugs
Gut bacteria generally exert reducing and hydrolytic reactions [12]. Enzymes associated with gut microorganisms are oxydoreductases, hydrolases, transferases, and lyases [13], widely distributed in a variety of bacterial groups, although enzymes with high similarity between families can catalyze different chemical reactions.
The gut microbiome metabolizes non-digestible dietary components [14] and participates in immune function [15] and/or bioactivation of nutrients and vitamins. However, not all the microbial enzymes involved in these chemical transformations have been described. Indeed, not all metabolic activities or functional profiles in these microbial communities can be predicted in metabolomic studies [16]. Thus, metabolic functions are assigned to superfamilies that can catalyze several different chemical reactions. One of the more interesting abilities of intestinal bacteria is the synthesis of metabolites produced directly by the gut microbiota or by the metabolism of dietary components, such as trimethylamine N-oxide (TMAO), indoxyl sulfate (IS), and indole-3 acetic acid (IAA) [17].
Trimethylamine N-Oxide (TMAO)
Concerning metabolites, intestinal bacteria metabolize part of the main group of phosphatidylcholine, in the metabolic via phosphatidylcholine-choline, producing an intermediate compound known as trimethylamine (TMA). TMA is generated from dietary compounds such as betaine, L-carnitine, and its metabolite γ-butyrobetaine (GBB), choline, and other choline-containing compounds [18].
Choline is an essential nutrient, contributing to neurotransmission, methyl transfer events, and cell membrane function. It is found in high quantities in food stuffs such as peanuts, eggs, meat, beef liver, and cauliflower [19]. It is obtained as free choline from foods of animal origin or as part of various compounds such as phosphocholine, phosphatidylcholine, and sphingomyelin. Free choline is absorbed throughout the small intestine and it is integrated into cell membranes or absorbed by the liver, where it can be converted to lecithin, phosphocholine, or betaine [20]. If the amount of choline exceeds the absorption capacity, it passes to the large intestine where it is metabolized by microbial action to methylamines [21] (Figure 1).
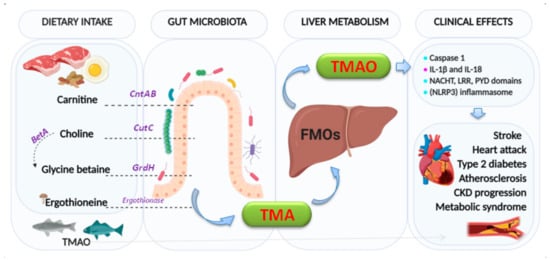
Figure 1. Formation of trimethylamine (TMA) and trimethylamine N-oxide reductase (TMAO). The key enzymes responsible indicated are: carnitine monooxygenase (CntAB); choline-TMA lyase (CutCD); glycine betaine reductase (GrdH); and Ergothionase. Choline to glycine betaine is mediated by the Bet pathway (BetA). FMOs: flavin-dependent monooxygenase (isoforms 1,3). Clinical effects:  : activation,
: activation,  : producing and secretion.
: producing and secretion.
On the other hand, L-carnitine is converted into TMAO by the action of carnitine oxidoreductase. Next, TMA is rapidly oxidized by hepatic flavin-containing monooxygenases (FMO1–FMO3) to form TMAO [22]. Figure 2 summarizes the principal pathways and enzymes involved in the production of TMA and TMAO.
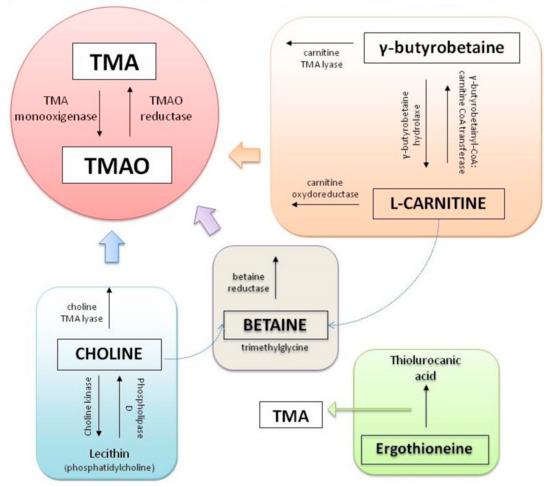
Figure 2. Principal pathways to formation of trimethylamine (TMA) and trimethylamine N-oxide reductase (TMAO).
There is a recognized association between TMAO and inflammation [23][24][25][26][27]. Indeed, TMAO triggers vascular inflammation by activation of caspase 1, producing and secreting pro-inflammatory cytokines (IL-1β and IL-18) and activating the NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome [28]. Activation of inflammasome has been linked to heart failure, adverse heart remodeling, and cytokine-mediated systolic dysfunction. The effects of TMAO on the components of the intrinsic cardiac autonomic nervous system (Ganglionated Plexi (GP) [29]) have been associated with increased expression via pro-inflammatory signaling and the nerve growth factors [28]. Whether circulating TMAO derived from the intrinsic microbiome can reach the GPs or not by creating sufficient local concentrations to give rise to arrhythmogenic effects [30] remains to be confirmed.
Likewise, elevated levels of TMAO in plasma increase the risk of developing atherosclerosis [31] and other adverse cardiovascular events, such as stroke, myocardial infarction, and death [22]. Recent studies in primary human coronary artery endothelial cells (HCAECs) isolated from normal human coronary arteries demonstrated that TMAO promoted nuclear translocation of nuclear factor-κB (NF-κB) and expression of tissue factor (TF), implicated in the thrombogenicity of atherosclerotic plaque [32]. Low-dose TMAO also significantly promoted low-dose of tumor necrosis factor-alpha (TNF-α) [33] or high mobility group box 1 (HMGB1) mediated by TF expression via activating NF-κB signaling and finally, studies in ST-elevation myocardial infarction patients, linking increased plasma concentrations TMAO with increased TF activity [34].
This entry is adapted from the peer-reviewed paper 10.3390/jcm10040715
References
- McIntyre, W.F.; Healey, J.S. Stroke Prevention for Patients with Atrial Fibrillation: Beyond the Guidelines. J. Atr. Fibrillation. 2017, 9.
- Lip, G.Y.; Banerjee, A.; Boriani, G.; Chiang, C.E.; Fargo, R.; Freedman, B.; Lane, D.A.; Ruff, C.T.; Turakhia, M.; Werring, D.; et al. Antithrombotic Therapy for Atrial Fibrillation. Chest 2018, 154, 1121–1201.
- Kirchhof, P.; Benussi, S.; Kotecha, D.; Ahlsson, A.; Atar, D.; Casadei, B.; Castella, M.; Diener, H.; Heidbuchel, H.; Hendriks, J.; et al. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur. Heart. J. 2016, 37, 2893–2962.
- Boriani, G.; Proietti, M.; Laroche, C.; Fauchier, L.; Marin, F.; Nabauer, M.; Potpara, T.; Dan, G.-A.; Kalarus, Z.; Diemberger, I.; et al. Contemporary stroke prevention strategies in 11 096 European patients with atrial fibrillation: A report from the EURObservational Research Programme on Atrial Fibrillation (EORP-AF) Long-Term General Registry. Europace 2018, 20, 747–757.
- Pengo, V.; Denas, G.; Zoppellaro, G.; Jose, S.P.; Hoxha, A.; Ruffatti, A.; Andreoli, L.; Tincani, A.; Cenci, C.; Prisco, D.; et al. Rivaroxaban vs warfarin in high-risk patients with antiphospholipid syndrome. Blood 2018, 132, 1365–1371.
- Limper, M.; De Leeuw, K.; Lely, A.T.; Westerink, J.; Teng, Y.K.O.; Eikenboom, J.; Otter, S.; Jansen, A.J.G.; Ree, M.V.D.; Spierings, J.; et al. Diagnosing and treating antiphospholipid syndrome: A consensus paper. Neth. J. Med. 2019, 77, 98–108.
- Tritschler, T.; Castellucci, L.A. It’s time for head-to-head trials with direct oral anticoagulants. Thromb. Res. 2019, 180, 64–69.
- Bochenek, T.; Czarnogorski, M.; Nizankowski, R.; Pilc, A. Are pharmacological properties of anticoagulants reflected in pharmaceutical pricing and reimbursement policy? Out-patient treatment of venous thromboembolism and utilization of anticoagulants in Poland. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 1649–1656.
- Rivera-Caravaca, J.M.; Roldán, V.; Esteve-Pastor, M.A.; Valdés, M.; Vicente, V.; Marín, F.; Lip, G.Y. Reduced Time in Therapeutic Range and Higher Mortality in Atrial Fibrillation Patients Taking Acenocoumarol. Clin. Ther. 2018, 40, 114–122.
- Kazemian, N.; Mahmoudi, M.; Halperin, F.; Wu, J.C.; Pakpour, S. Gut microbiota and cardiovascular disease: Opportunities and challenges. Microbiome 2020, 8, 1–17.
- Ahmad, A.F.; Dwivedi, G.; O’Gara, F.; Caparros-Martin, J.; Ward, N.C. The gut microbiome and cardiovascular disease: Current knowledge and clinical potential. Am. J. Physiol. Circ. Physiol. 2019, 317, H923–H938.
- Sousa, T.; Paterson, R.; Moore, V.; Carlsson, A.; Abrahamsson, B.; Basit, A.W. The gastrointestinal microbiota as a site for the biotransformation of drugs. Int. J. Pharm. 2008, 363, 1–25.
- Linhardt, R.J.; Galliher, P.M.; Cooney, C.L. Polysaccharide lyases. Appl. Biochem. Biotechnol. 1986, 12, 135–176.
- Vernocchi, P.; Del Chierico, F.; Putignani, L. Gut Microbiota Metabolism and Interaction with Food Components. Int. J. Mol. Sci. 2020, 21, 3688.
- Fung, T.C.; Olson, C.A.; Hsiao, T.C.F.C.A.O.E.Y. Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 2017, 20, 145–155.
- Zimmermann, J.; Kaleta, C.; Waschina, S. gapseq: Informed prediction of bacterial metabolic pathways and reconstruction of accurate metabolic models. bioRxiv 2020.
- Fernandez-Prado, R.; Esteras, R.; Perez-Gomez, M.V.; Gracia-Iguacel, C.; Gonzalez-Parra, E.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Nutrients Turned into Toxins: Microbiota Modulation of Nutrient Properties in Chronic Kidney Disease. Nutrients 2017, 9, 489.
- Janeiro, M.H.; Ramírez, M.J.; Milagro, F.I.; Martínez, J.A.; Solas, M. Implication of Trimethylamine N-Oxide (TMAO) in Disease: Potential Biomarker or New Therapeutic Target. Nutrients 2018, 10, 1398.
- Zeisel, S.H.; Mar, M.-H.; Howe, J.C.; Holden, J.M. Concentrations of Choline-Containing Compounds and Betaine in Common Foods. J. Nutr. 2003, 133, 1302–1307.
- Zeisel, S.H. Choline deficiency. J. Nutr. Biochem. 1990, 1, 332–349.
- Zeisel, S.H.; Wishnok, J.S.; Blusztajn, J.K. Formation of methylamines from ingested choline and lecithin. J. Pharmacol. Exp. Ther. 1983, 225, 320–324.
- Zeisel, S.H.; Warrier, M. TrimethylamineN-Oxide, the Microbiome, and Heart and Kidney Disease. Annu. Rev. Nutr. 2017, 37, 157–181.
- Chen, K.; Zheng, X.; Feng, M.; Li, D.; Zhang, H. Gut Microbiota-Dependent Metabolite Trimethylamine N-Oxide Contributes to Cardiac Dysfunction in Western Diet-Induced Obese Mice. Front. Physiol. 2017, 8, 139.
- Chen, M.L.; Zhu, X.H.; Ran, L.; Lang, H.D.; Yi, L.; Mi, M.T. Trimethylamine-N-Oxide Induces Vascular Inflammation by Activating the NLRP3 Inflammasome Through the SIRT3-SOD2-mtROS Signaling Pathway. J. Am. Heart Assoc. 2017, 6, e006347.
- Rohrmann, S.; Linseisen, J.; Allenspach, M.; Von Eckardstein, A.; Müller, D. Plasma Concentrations of Trimethylamine-N-oxide Are Directly Associated with Dairy Food Consumption and Low-Grade Inflammation in a German Adult Population. J. Nutr. 2016, 146, 283–289.
- Sun, X.; Jiao, X.; Ma, Y.; Liu, Y.; Zhang, L.; He, Y.; Chen, Y. Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ROS-TXNIP-NLRP3 inflammasome. Biochem. Biophys. Res. Commun. 2016, 481, 63–70.
- Yue, C.; Yang, X.; Li, J.; Chen, X.; Zhao, X.; Chen, Y.; Wen, Y. Trimethylamine N-oxide prime NLRP3 inflammasome via inhibiting ATG16L1-induced autophagy in colonic epithelial cells. Biochem. Biophys. Res. Commun. 2017, 490, 541–551.
- Heianza, Y.; Ma, W.; Manson, J.E.; Rexrode, K.M.; Qi, L. Gut Microbiota Metabolites and Risk of Major Adverse Cardiovascular Disease Events and Death: A Systematic Review and Meta-Analysis of Prospective Studies. J. Am. Hear. Assoc. 2017, 6.
- Lau, D.H.; Linz, D.; Schotten, U.; Mahajan, R.; Sanders, P.; Kalman, J.M. Pathophysiology of Paroxysmal and Persistent Atrial Fibrillation: Rotors, Foci and Fibrosis. Hear. Lung Circ. 2017, 26, 887–893.
- Mishima, R.S.; Elliott, A.D.; Sanders, P.; Linz, D. Microbiome and atrial fibrillation. Int. J. Cardiol. 2018, 255, 103–104.
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.A.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.-M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nat. Cell Biol. 2011, 472, 57–63.
- Cheng, X.; Qiu, X.; Liu, Y.; Yuan, C.; Yang, X. Trimethylamine N-oxide promotes tissue factor expression and activity in vascular endothelial cells: A new link between trimethylamine N-oxide and atherosclerotic thrombosis. Thromb. Res. 2019, 177, 110–116.
- Zhu, Y.; Li, Q.; Jiang, H. Gut microbiota in atherosclerosis: Focus on trimethylamine N-oxide. APMIS. 2020, 128, 353–366.
- Subramaniam, S.; Boukhlouf, S.; Fletcher, C. A bacterial metabolite, trimethylamine N-oxide, disrupts the hemostasis balance in human primary endothelial cells but no coagulopathy in mice. Blood Coagul. Fibrinolysis. 2019, 30, 324–330.