Pediatric ependymoma (EPN) is a highly aggressive tumor of the central nervous system that remains incurable in 40% of cases. In children, the majority of cases develop in the posterior fossa and can be classified into two distinct molecular entities: EPN posterior fossa A (PF-EPN-A) and EPN posterior fossa B (PF-EPN-B). Patients with PF-EPN-A have poor outcome and are in demand of new therapies. In general, PF-EPN-A tumors show a balanced chromosome copy number profile and have no recurrent somatic nucleotide variants. However, these tumors present abundant epigenetic deregulations, thereby suggesting that epigenetic therapies could provide new opportunities for PF-EPN-A patients. In vitro epigenetic drug screening of 11 compounds showed that histone deacetylase inhibitors (HDACi) had the highest anti-proliferative activity in two PF-EPN-A patient-derived cell lines. Further screening of 5 new brain-penetrating HDACi showed that CN133 induced apoptosis in vitro, reduced tumor growth in vivo and significantly extended the survival of mice with orthotopically-implanted EPN tumors by modulation of the unfolded protein response, PI3K/Akt/mTOR signaling, and apoptotic pathways among others.
- pediatric brain tumors
- posterior fossa ependymoma
- epigenetic therapies
- histone deacetylase inhibitors (HDACi)
1. Introduction
Pediatric tumors of the central nervous system (CNS) are the most common solid tumors in children, second only to hematologic malignancies and are the leading cause of cancer-related deaths [1]. Despite all current therapeutic efforts, almost 50–60% of patients will not achieve a long-term cure owing to disease progression and resistance to existing therapies. This is particularly true for ependymomas (EPNs), the third most common pediatric brain tumors after astrocytomas and medulloblastomas [2].
EPNs are very heterogeneous in terms of age at diagnosis, molecular profile, histologic grade, and clinical behavior and can appear along the entire craniospinal axis, most commonly in the posterior fossa, supratentorial, and spinal cord (reviewed in [3]). Recent genomic studies based on DNA methylation profiles identified nine distinct molecular subgroups, three within each CNS compartment [4]. Among them, the most aggressive ones are RELA fusion-positive supratentorial EPN (ST-EPN-RELA) and posterior fossa EPN group A (PF-EPN-A) [3]. While for ST-EPN-RELA tumors, the identification of the driver oncogene and the mapping of the molecular associated alterations may facilitate the roadmap for therapeutic strategies [5][6], PF-EPN-A tumors are more heterogeneous and with low mutation rate, and no significant recurrent somatic single nucleotide variants [4][7]. However, a poor-prognosis subset of these tumors exhibits a CpG island methylate phenotype [8], which suggests that could be sensitive to epigenetic modifiers, such as polycomb repressor complex 2 (PRC2) inhibition, DNA methylation inhibitors, and histone deacetylase (HDAC) inhibitors, either alone or in combination [9].
Since a complete resection of these tumors is almost impossible and no benefits of current chemotherapies have been observed [10], in this study, we sought to screen the therapeutic potential of epigenetic drugs in PF-EPN-A models. Among them, histone deacetylase inhibitors (HDACi) were found to have the greatest anti-proliferative activity. The addition of acetyl groups from acetyl-CoA to specific lysine residues is controlled by histone acetyl-transferases (HATs) associated generally with active transcription, and removed by histone deacetylases (HDACs), which is generally connected with transcriptional repression [11].
To date, many HDAC inhibitors (HDACis) have been tested in oncology clinical trials, which led to the approval of Vorinostat, Romidepsin, Bellinostat, and Panobinostat for the treatment of cutaneous and peripheral T-cell lymphoma and multiple myeloma [12]. In brain tumors, however, tolerable doses of the HDACi Vorinostat showed no anti-tumoral effects, suggesting that it did not reach the brain at therapeutic doses [13]. Additional studies have clearly demonstrated that Vorinostat has a poor brain penetrability [14]. Owing to the large therapeutic potential of HDACis already observed in non-brain tumors, it would be desirable to find new HDACis able to reach the brain at therapeutic doses for the treatment of CNS malignancies.
Therefore, we analyzed the therapeutic potential of 5 novel brain-penetrating HDACis and found the CN133 compound to be a potent inductor of apoptotic cell death in vitro and capable of reducing tumor growth in vivo. Of note, CN133 is capable of inducing histone acetylation in orthotopically-implanted PF-EPN-A tumors at similar levels to other peripheral tissues such as the spleen showing a much better targeting of histones than other HDACis currently in clinical use. In summary, our results provide strong preclinical evidence for the use of this new HDACi, with unique brain-penetrating and safety profile, for the treatment of PF-EPN-A tumors.
2. CN133 and CN147 HDACis Induce Apoptosis in EPN Cell Lines
For better understanding how CN133 and CN147 affect cell viability, flow cytometry analyses were performed. CN133 and CN147 induced a significant increase in the percentage of cells in the sub-G0/G1 peak (Figure 1a–c), one of the hallmarks of apoptotic cell death. Furthermore, an increase in the amount of the cleaved and active form of caspase-3 (i.e., 17 kD fragment) and cleavage of the caspase-3 substrate PARP were also observed. Next, we analyzed another apoptotic hallmark, i.e., the exposition of phosphatidylserine in the outer membrane of apoptotic cells, a fact that can be detected with anti-annexin V antibodies. Compared to DMSO-treated cells, CN133 and CN147 increased the percentage of cells that were annexin V positive, either negative for propidium iodide (commonly referred as early apoptosis) or positive for both markers (i.e., late apoptosis). The percentage of cells only positive for propidium iodide (considered necrotic cells) did not vary significantly (Figure 1d,e). In summary, these analyses confirmed that both compounds trigger apoptotic cell death in EPN cell lines.
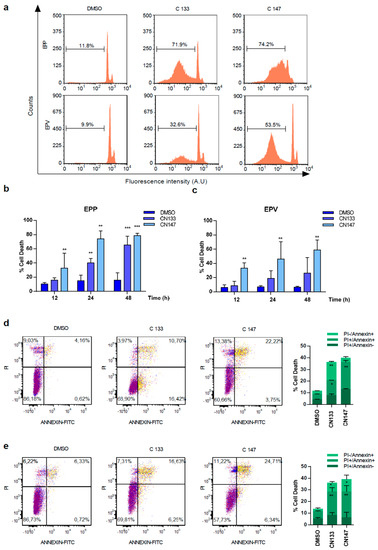
Figure 1. CN133 and CN147 induce cell death in EPN cell lines: (a) EPP and EPV cell lines were treated with 1 μM of CN133 and CN147 and apoptosis was evaluated by flow cytometry (FACS); (b,c) Graph represents the average quantification of the percentage of cells in sub-G1 of three independent experiments (n = 3/experiment) ± SEM. ** p < 0.01; *** p < 0.001; EPP (d) and EPV (e) were treated with 1 µM of CN133 and CN147 for 24 h. At the end of the treatment, cells were harvested and stained with the Annexin V kit following the manufacturer’s instruction. Graph represent the average quantification (n = 3/each). * p < 0.05; ** p < 0.01; *** p < 0.001.
3. CN133 Impairs Tumour Growth and Extends Survival of Mice Bearing EPN Orthotopic Xenografts
To understand how CN133 and CN147 modulate histone acetylation in EPN cell lines and predict potential behavior in vivo, both compounds were administered in a “constant” compared with a “pulse” setting. Under both conditions, HDACi treatments induced a rapid increase in the acetylation of different histone residues, such as H2BK5, H3-N-term, and H3K9Me2. The hyperacetylation at all marks reached maximum levels at 4 h and remained constant as long as the inhibitor was present in the cell media. However, when EPP cell lines were treated for 6 h, washed and further incubated for up to 18–24 h in inhibitor-free media, the histone acetylation levels returned to basal levels. These results suggest that the compounds hit the targets in a transient and reversible manner. Thus, repetitive treatments would be needed to maintain high histone acetylation levels in vivo.
Next, to identify possible adverse effects resulting from exposure to these compounds in vivo, six-week-old NMRI nude mice were treated i.p. daily with either 1 mg/kg or 5 mg/kg for 1 week either once or twice a day. Average mouse weight showed no major differences between the control and compound-treated groups with either one or two injections per day (Figure 2a,b).
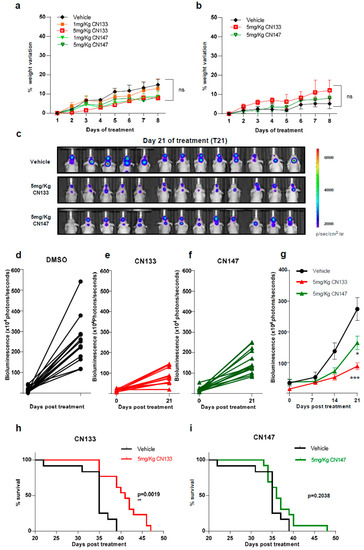
Figure 2. CN133 delays EPN tumors growth and extends survival of mice bearing EPN orthotopic xenografts. Percentage of weight variation of nude mice treated with 1 or 5 mg/kg of CN133 or CN147 for 8 days by i.p. administration (n = 5 mice/group) once (a) or twice a day (b); (c) representative bioluminescent images at the indicated days of EPP luciferase-transduced cells orthotopically-injected in the posterior fossa of nude mice; (d–f) bioluminescence quantification of EPP tumors progression at 21 days post treatment DMSO (d) C133 (e) and C147 (f); (g) bioluminescence quantification of EPP tumors over time (h,i) Kaplan–Meier survival curves of mice implanted with EPP luciferase-transduced cells (n = 13/group) and treated with vehicle or the indicated drugs.
To determine the anti-tumoral efficacy of CN133 and CN147 in vivo, EPN orthotopic xenografts were established by stereotaxic implantation of luciferase-expressing EPN cells into the fourth ventricle of nude mice. Cells were left to engraft and form a tumor for a period of 10 days and then, mice were randomized into three groups, namely, vehicle (n = 13), CN133 (n = 13), and CN147 (n = 13), and treated twice a day for 40 days. Tumor growth was monitored by in vivo bioluminescent imaging once a week. While tumors of vehicle-treated mice grew very fast, those tumors of HDACi-treated grew at a much slower pace (Figure 2c–f). Statistically significant differences were appreciated starting day 21 of treatment, with CN133 being more effective than CN147 (Figure 2g). However, only CN133 significantly prolonged the animal’s lifespan (p = 0.0019, Figure 2h,i). Collectively, these data suggest that CN133 exerts a better therapeutic effect than CN147 on EPN in vivo.
Finally, to confirm that CN133 was able to cross the blood–brain barrier and target HDACs in the implanted tumor cells more effectively than the other HDACis, mice were treated for 14 days with either 5 mg/kg CN133 or 50 mg/kg SAHA (Vorinostat), the most commonly-used HDACi for the treatment of brain tumors. A peripheral tissue (i.e., spleen) and brain tumors were collected 4 h after the last drug administration and histone acetylation was analyzed. H2B resulted dramatically hyperacetylated at residue H2BK5Ac in both spleen and brain tumors of CN133-treated mice, while SAHA treatment only mildly affected H2B acetylation levels (Figure 3a–d). Of note, H2B hyperacetylation levels induced by CN133 were very similar in EPN tumors and spleen, which further supports the good brain penetrability profile of CN133. These findings corroborate the evidence that CN133 is a much more potent HDACi than Vorinostat in increasing histone acetylation in brain tumors, and places CN133 as a new promising therapeutic candidate for ependymoma treatment.
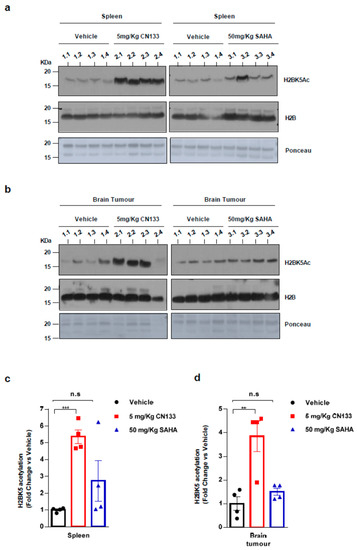
Figure 3. CN133 is more potent than SAHA in increasing histone acetylation in vivo: Western Blot of spleen (a) or brain tumors (b) from mice treated for 14 days with 5 mg/kg C133 or 50 mg/kg SAHA administered twice a day by i.p. Numbers above the western blots indicate mouse identification. Uncropped Blots of Figure 3a,b are shown in Figure S9 (c,d) Densitometry analysis of H2BK5ac levels normalized to H3. Fold changes are relative to values from vehicle-injected mice. ** p < 0.01; *** p < 0.001.
4. Discussion
Recent genomic and transcriptional analyses showed that the most aggressive and frequent subtypes of EPN, such as PF-EPN-A, harbor very few recurrent genetic alterations while possessing a significant proportion of frequent events converging on epigenetic mechanisms [15]. These findings have may open up new avenues for PF-EPN-A therapy by targeting epigenetic modifications.
Several clinical trials on pediatric patients showed epigenetic drugs to be safe and well tolerated [16][17][18]. The present study sought to analyses the therapeutic potential of preclinical-validated epigenetic drugs known to exhibit antitumor activity in brain tumor models and identified targets (i.e., histone-lysine methyltransferase inhibitors, bromodomain inhibitors, and HDACis). This screening performed in patient-derived PF-EPN-A cell lines showed HDACis to be the epigenetic compounds with the highest therapeutic potential.
In recent years, altered expression and mutations of genes encoding for HDACs have been linked to tumor development since, together with histone acetyltransferases, they concur to promote an aberrant transcription of key genes involved in controlling cell proliferation, cell-cycle regulation and apoptosis. Mining mRNA microarray expression data for ependymoma (GSE64415, n = 209) showed that HDAC1, HDAC2, and HDAC3 mRNA expression levels were significantly upregulated in EPN compared to CNS tissues (i.e., whole brain GSE11882, n = 172; cerebellum GSE3526, n = 9), thus underlining a potential role for these enzymes in the development of this particular type of pediatric brain tumor. The anti-tumor efficacy of pan-specific HDACis was investigated and promising results were obtained by treating patients with some FDA-approved drugs, such as Vorinostat, Romidepsin, Bellinostat, and Panobinostat. In particular, Vorinostat has been approved for the treatment of cutaneous T-cell lymphoma, and Panobinostat for multiple myeloma. Despite promising preclinical evidence in EPN models [19], HDACis did not yield the expected clinical results in patients with CNS tumors, most probably due to their inability to reach and accumulate in the brain at the concentration required to elicit a therapeutic response [20].
Here, five new HDACis proved to impact on PF-EPN-A cell lines viability even more robustly than the FDA-approved Vorinostat. All compounds were able to strongly enhance the acetylation levels of Histone H3, CN133 and CN147 being the most effective. Interestingly, pharmacokinetics and brain distribution analyses revealed that both compounds are characterized by an excellent brain accumulation profile, reaching brain-to-plasma ration of 20:1 upon intraperitoneal administration. Furthermore, they demonstrated strong potency and isozyme selectivity for HDAC1, HDAC2, HDAC3, HDAC4, HDAC5, and HDAC6 in a recombinant human enzyme assay.
When these compounds were used to treat orthotopically-implanted PF-EPN-A xenografts, both CN133 and CN147 significantly delayed tumor growth, although only CN133 was able to prolong the lifespan of treated animals. We believe these differences were not linked to the ability of the compounds to reach and accumulate in the brain, since both showed the same brain-to-plasma partition of 20:1, but to a difference in potency to inhibit HDACs. In fact, CN133 exhibited an IC50 for HDAC1, 2, and 3, which is almost 500 times lower as compared to that of CN147. Moreover, while CN133 reduced EPN cell viability in dose-dependent manner, CN147 showed cytotoxic activity when overcoming a specific concentration threshold, kinetic behavior quite difficult to control in the in vivo scenario.
Understanding how HDACis exert their therapeutic effect is complex. First, HDACs associates with distinct multi-subunit complexes which exhibit diverse, and often, cell type-specific functions. These HDAC interacting partners not only modulate the catalytic activity and substrate specificity of the histone deacetylase(s) present in the multiprotein complex, but also alter the accessibility of catalytic sites of HDACs for the inhibitor itself. Second, HDACs can also interact with each other, adding further complexity to interaction network. Third, although the name HDAC implies some specificity for histones, HDACs are known to affect the acetylation status of a wide variety of non-histone proteins, including transcription factors and chaperones, among others, resulting in changes in protein stability, protein-protein, and protein-DNA interactions [21].
Transcriptome and gene set enrichment analyses showed that the CN133 compound modulates several cell-signaling pathways in EPN cells which ultimately lead to cell cycle arrest and apoptosis. In particular, the cyclin-dependent kinase inhibitor p21 (CDKN1A) appears to be one of the most robustly and rapidly up-regulated genes. p21 overexpression may lead to cell cycle arrest in G1 phase and induction of apoptosis (reviewed in [22]), a mechanism shown to occur in other brain tumor cell lines treated with different HDACis, such as Vorinostat, Trichostatin A [23], Entinostat [24], Dacinostat, or Sodium butyrate [25]. Although the activation of p53 usually may be responsible for the upregulation of p21 in response to drug treatments, different mechanisms have been proposed to be responsible for the upregulation of p21 upon HDACi treatment. In particular, Richon et al. reported that HDACis, such as SAHA, were able to increase the acetylation of histones at specific genomic sites, such as the p21 locus, thereby inducing the direct transcription of p21 [26] in a p53 independent manner. Our evidence supports this mechanism, since CN133 is able to increase p21 even when p53 is silenced, or in other cell lines with non-functional p53. CN133 also negatively impacts on the PI3K/AKT/mTOR pathway, as evidenced by the reduction in phosphorylated levels of AKTS473 and the downstream target ribosomal protein S6. Similar results have been observed in B-lymphoma cells treated with the HDACi MPT0E028 [27].
Moreover, CN133 also modulates the expression of several factors of the unfolded protein response (UPR) pathway. The CN133-dependent induction of UPR could be mediated by modulation of hypoxia-related genes (i.e., HIF-1α) and/or by the direct acetylation of glucose-regulated protein 78 (GRP78), a chaperone molecule assisting the folding, assembly, and translocation of newly-synthesized polypeptides across the ER membrane. During ER stress, the dramatic increase in unfolded substrates leads to the sequestration of GRP78, releasing sensors, such as PERK to initiate UPR signals [28][29]. ER-stress activation mechanism leading to apoptotic cell death has also been observed in neuroblastoma cells treated with SAHA [28]. UPR-related proteins, such as ATF3, ATF4, and CHOP were upregulated at 24 h post-treatment, a phenomenon that could be sufficient to trigger apoptotic cell death. In fact, downregulation of anti-apoptotic protein XIAP at earlier time points and the concomitant upregulation of the pro-apoptotic protein NOXA, would confirm orchestrated induction of the apoptotic cascade. A similar chain of events has been described for glioma or neuroblastoma cells treated with HDACi [30][31].
Although an optimization of drug dosing and administration schedules could significantly improve the therapeutic potential of CN133 as a monotherapy, a combination of epigenetic compounds with standard drugs or radiation regimens is often proposed in clinical trials [32]. For example, the combination of the HDACi Vorinostat and Valproic acid with TMZ and/or radiotherapy in CNS malignancies such as diffuse intrinsic pontine glioma (DIPG) or adult GBM improved the outcomes in the patients with a 1-year OS rate of 86% (CI: 76–98) and a 6-month PFS rate of 70% (CI: 57–87) [33]. Thus, the use of CN133 in combinations with standard therapies could improve the patient’s outcome, a fact that warrants further investigation.
This entry is adapted from the peer-reviewed paper 10.3390/cancers12071922
References
- Gatta, G.; Botta, L.; Rossi, S.; Aareleid, T.; Bielska-Lasota, M.; Clavel, J.; Dimitrova, N.; Jakab, Z.; Kaatsch, P.; Lacour, B.; et al. Childhood cancer survival in Europe 1999–2007: Results of EUROCARE-5—A population-based study. Lancet Oncol. 2014, 15, 35–47.
- Cage, T.A.; Clark, A.J.; Aranda, D.; Gupta, N.; Sun, P.P.; Parsa, A.T.; Auguste, K.I. A systematic review of treatment outcomes in pediatric patients with intracranial ependymomas. J. Neurosurg. Pediatr. 2013, 11, 673–681.
- Pajtler, K.W.; Pfister, S.M.; Kool, M. Molecular dissection of ependymomas. Oncoscience 2015, 2, 827–828.
- Pajtler, K.W.; Witt, H.; Sill, M.; Jones, D.T.; Hovestadt, V.; Kratochwil, F.; Wani, K.; Tatevossian, R.; Punchihewa, C.; Johann, P.; et al. Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 2015, 27, 728–743.
- Parker, M.; Mohankumar, K.M.; Punchihewa, C.; Weinlich, R.; Dalton, J.D.; Li, Y.; Lee, R.; Tatevossian, R.G.; Phoenix, T.N.; Thiruvenkatam, R.; et al. C11orf95-RELA fusions drive oncogenic NF-kappaB signalling in ependymoma. Nature 2014, 506, 451–455.
- Ozawa, T.; Arora, S.; Szulzewsky, F.; Juric-Sekhar, G.; Miyajima, Y.; Bolouri, H.; Yasui, Y.; Barber, J.; Kupp, R.; Dalton, J.; et al. A De Novo Mouse Model of C11orf95-RELA Fusion-Driven Ependymoma Identifies Driver Functions in Addition to NF-kappaB. Cell Rep. 2018, 23, 3787–3797.
- Witt, H.; Mack, S.C.; Ryzhova, M.; Bender, S.; Sill, M.; Isserlin, R.; Benner, A.; Hielscher, T.; Milde, T.; Remke, M.; et al. Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell 2011, 20, 143–157.
- Mack, S.C.; Witt, H.; Piro, R.M.; Gu, L.; Zuyderduyn, S.; Stutz, A.M.; Wang, X.; Gallo, M.; Garzia, L.; Zayne, K.; et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 2014, 506, 445–450.
- Ramaswamy, V.; Taylor, M.D. Treatment implications of posterior fossa ependymoma subgroups. Chin. J. Cancer 2016, 35, 93.
- Thorp, N.; Gandola, L. Management of Ependymoma in Children, Adolescents and Young Adults. Clin. Oncol. (R. Coll. Radiol.) 2019, 31, 162–170.
- Yang, X.J.; Seto, E. HATs and HDACs: From structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007, 26, 5310–5318.
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 8, 1414.
- Galanis, E.; Jaeckle, K.A.; Maurer, M.J.; Reid, J.M.; Ames, M.M.; Hardwick, J.S.; Reilly, J.F.; Loboda, A.; Nebozhyn, M.; Fantin, V.R.; et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: A north central cancer treatment group study. J. Clin. Oncol. 2009, 27, 2052–2058.
- Hanson, J.E.; La, H.; Plise, E.; Chen, Y.H.; Ding, X.; Hanania, T.; Sabath, E.V.; Alexandrov, V.; Brunner, D.; Leahy, E.; et al. SAHA enhances synaptic function and plasticity in vitro but has limited brain availability in vivo and does not impact cognition. PLoS ONE 2013, 8, e69964.
- Tran, T.H.; Shah, A.T.; Loh, M.L. Precision Medicine in Pediatric Oncology: Translating Genomic Discoveries into Optimized Therapies. Clin. Cancer Res. 2017, 23, 5329–5338.
- Gore, L.; Triche, T.J., Jr.; Farrar, J.E.; Wai, D.; Legendre, C.; Gooden, G.C.; Liang, W.S.; Carpten, J.; Lee, D.; Alvaro, F.; et al. A multicenter, randomized study of decitabine as epigenetic priming with induction chemotherapy in children with AML. Clin. Epigenetics 2017, 9, 108.
- Italiano, A.; Soria, J.C.; Toulmonde, M.; Michot, J.M.; Lucchesi, C.; Varga, A.; Coindre, J.M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018, 19, 649–659.
- Karahoca, M.; Momparler, R.L. Pharmacokinetic and pharmacodynamic analysis of 5-aza-2′-deoxycytidine (decitabine) in the design of its dose-schedule for cancer therapy. Clin. Epigenetics 2013, 5, 3.
- Milde, T.; Kleber, S.; Korshunov, A.; Witt, H.; Hielscher, T.; Koch, P.; Kopp, H.G.; Jugold, M.; Deubzer, H.E.; Oehme, I.; et al. A novel human high-risk ependymoma stem cell model reveals the differentiation-inducing potential of the histone deacetylase inhibitor Vorinostat. Acta Neuropathol. 2011, 122, 637–650.
- Lee, P.; Murphy, B.; Miller, R.; Menon, V.; Banik, N.L.; Giglio, P.; Lindhorst, S.M.; Varma, A.K.; Vandergrift, W.A., 3rd; Patel, S.J.; et al. Mechanisms and clinical significance of histone deacetylase inhibitors: Epigenetic glioblastoma therapy. Anticancer Res. 2015, 35, 615–625.
- Reichert, N.; Choukrallah, M.A.; Matthias, P. Multiple roles of class I HDACs in proliferation, differentiation, and development. Cell Mol. Life Sci. 2012, 69, 2173–2187.
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414.
- Komata, T.; Kanzawa, T.; Nashimoto, T.; Aoki, H.; Endo, S.; Kon, T.; Takahashi, H.; Kondo, S.; Tanaka, R. Histone deacetylase inhibitors, N-butyric acid and trichostatin A, induce caspase-8- but not caspase-9-dependent apoptosis in human malignant glioma cells. Int. J. Oncol. 2005, 26, 1345–1352.
- Bangert, A.; Hacker, S.; Cristofanon, S.; Debatin, K.M.; Fulda, S. Chemosensitization of glioblastoma cells by the histone deacetylase inhibitor MS275. Anticancer Drugs 2011, 22, 494–499.
- Egler, V.; Korur, S.; Failly, M.; Boulay, J.L.; Imber, R.; Lino, M.M.; Merlo, A. Histone deacetylase inhibition and blockade of the glycolytic pathway synergistically induce glioblastoma cell death. Clin. Cancer Res. 2008, 14, 3132–3140.
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. USA 2000, 97, 10014–10019.
- Huang, H.L.; Peng, C.Y.; Lai, M.J.; Chen, C.H.; Lee, H.Y.; Wang, J.C.; Liou, J.P.; Pan, S.L.; Teng, C.M. Novel oral histone deacetylase inhibitor, MPT0E028, displays potent growth-inhibitory activity against human B-cell lymphoma in vitro and in vivo. Oncotarget 2015, 6, 4976–4991.
- Chen, Y.; Tsai, Y.H.; Tseng, S.H. RECK regulated endoplasmic reticulum stress response and enhanced cisplatin-induced cell death in neuroblastoma cells. Surgery 2013, 154, 968–979.
- Chueh, A.C.; Tse, J.W.T.; Dickinson, M.; Ioannidis, P.; Jenkins, L.; Togel, L.; Tan, B.; Luk, I.; Davalos-Salas, M.; Nightingale, R.; et al. ATF3 Repression of BCL-XL Determines Apoptotic Sensitivity to HDAC Inhibitors across Tumor Types. Clin. Cancer Res. 2017, 23, 5573–5584.
- Bangert, A.; Cristofanon, S.; Eckhardt, I.; Abhari, B.A.; Kolodziej, S.; Hacker, S.; Vellanki, S.H.; Lausen, J.; Debatin, K.M.; Fulda, S. Histone deacetylase inhibitors sensitize glioblastoma cells to TRAIL-induced apoptosis by c-myc-mediated downregulation of cFLIP. Oncogene 2012, 31, 4677–4688.
- Francisco, R.; Perez-Perarnau, A.; Cortes, C.; Gil, J.; Tauler, A.; Ambrosio, S. Histone deacetylase inhibition induces apoptosis and autophagy in human neuroblastoma cells. Cancer Lett. 2012, 318, 42–52.
- Munster, P.N.; Marchion, D.; Thomas, S.; Egorin, M.; Minton, S.; Springett, G.; Lee, J.H.; Simon, G.; Chiappori, A.; Sullivan, D.; et al. Phase I trial of vorinostat and doxorubicin in solid tumours: Histone deacetylase 2 expression as a predictive marker. Br. J. Cancer 2009, 101, 1044–1050.
- Krauze, A.V.; Myrehaug, S.D.; Chang, M.G.; Holdford, D.J.; Smith, S.; Shih, J.; Tofilon, P.J.; Fine, H.A.; Camphausen, K. A Phase 2 Study of Concurrent Radiation Therapy, Temozolomide, and the Histone Deacetylase Inhibitor Valproic Acid for Patients With Glioblastoma. Int. J. Radiat. Oncol. Biol. Phys. 2015, 92, 986–992.