A thorough overview of principles, recent technological breakthroughs, and current status of TCR gene therapy for cancer.
- adoptive cell therapy
- gene editing
- T-cell receptor engineering
- neoantigen targeting
1. Introduction
Immune checkpoint inhibitors have redefined the treatment of cancer, but their efficacy depends critically on the presence of sufficient tumor-specific lymphocytes, and cellular immunotherapies develop rapidly to fill this gap. The paucity of suitable extracellular and tumor-associated antigens in solid cancers necessitates the use of neoantigen-directed T-cell-receptor (TCR)-engineered cells, while prevention of tumor evasion requires combined targeting of multiple neoepitopes. These can be currently identified within 2 weeks by combining cutting-edge next-generation sequencing with bioinformatic pipelines and used to select tumor-reactive TCRs in a high-throughput manner for expeditious scalable non-viral gene editing of autologous or allogeneic lymphocytes. “Young” cells with a naive, memory stem or central memory phenotype can be additionally armored with “next-generation” features against exhaustion and the immunosuppressive tumor microenvironment, where they wander after reinfusion to attack heavily pretreated and hitherto hopeless neoplasms. Facilitated by major technological breakthroughs in critical manufacturing steps, based on a solid preclinical rationale, and backed by rapidly accumulating evidence, TCR therapies break one bottleneck after the other and hold the promise to become the next immuno-oncological revolution.
The advent of immunotherapy was a crucial advance for medical oncology: for the first time ever, five-year survival became feasible for patients with highly lethal solid tumors, such as advanced melanoma and non-small-cell lung carcinoma (NSCLC) [1][2][3][4]. Immune checkpoint inhibitors (ICI), mainly PD-(L)1 blockers, are meanwhile approved for the treatment of most metastatic human cancers, while favorable evidence accumulates rapidly for earlier-stage diseases, as well [5]. Nevertheless, a significant fraction of patients does not benefit from contemporary immuno-oncologic (IO) options, the activity of which critically depends on adequate numbers of tumor-specific T-cells being present in the host and especially in the tumor microenvironment (TME) [6]. Hence, adoptive cellular therapies (ACT) are the currently rising next IO wave, aiming to fill this gap. They represent the most rapidly expanding sector of modern cancer immunotherapy and comprised 31% of the entire IO pipeline as of August 2019 [7].
2. The Unique Potential of T-Cell-Receptor (TCR) Therapy
Historically, the first source of tumor-specific T cells have been the patient’s own tumor-infiltrating lymphocytes (TIL), which can be isolated from tumor tissue, expanded using cytokines together with feeder cells or antibody-coated beads, and reinfused after lymphodepleting conditioning with subsequent IL-2 support [1]. Although this approach has consistently shown response rates of up to 50% in pretreated melanoma patients [2] and is technically feasible for lung and other solid cancers, as well [3], it has one major drawback: most epithelial cancers are poorly immunogenic, so that only a tiny fraction of harvested lymphocytes are actually active against the tumor, and reinfusion of bulk TIL does not result in responses [4]. In order to improve outcome, tumor-reactive T cells can be selected among bulk TIL based on the surface expression of various markers, such as PD-1 or CD137, and/or the tumor’s neoantigenic profile. However, these pipelines are still too time-consuming for routine application, with turn-over times of several weeks, and the product quality remains impaired by the T-cell exhaustion induced already before harvesting in the tumor microenvironment (TME) [5][6].
Genetic engineering of T cells obtained from the patients’ blood by leukapheresis overcomes both problems. These can be transduced with antigen receptors directed against tumor antigens, either classical T-cell (TCR) or chimeric (CAR), expanded and reinfused in a similar manner [7]. Although CAR-T cells are currently more advanced in clinical development and already approved for the treatment of various CD19+ hematologic malignancies [8], TCR-T cells combine several important advantages and are expected to become the mainstay of ACT for solid tumors [9][10][11].
In the first place, the number of antigens amenable to TCR-based therapies is much higher than of those for CAR, as less than 25% of human proteins are membrane-bound, and a considerably lower fraction of all amino acid sequences (probably <10%) will be accessible on the cell surface [10][12] (Table 1). While CARs can recognize only extracellular proteins, glycoproteins, glycolipids or carbohydrates through their single-chain variable fragment (scFv) [13], the peptide-major histocompatibility complex (pMHC) combinations recognized by the TCR are drawn from both the intra- and extra-cellular compartment [14]. Moreover, TCRs require lower amounts of antigen for activation than CARs, (generally 1–50/cell vs. >103/cell), which is possibly linked to additional involvement of the CD4 or CD8 co-receptors [15], a higher number of immunoreceptor tyrosine-based activation motifs (10 vs. 3), and the ability of different TCR molecules to serially engage the antigens of low abundance, which amplifies responses [16][17][18][19]. At the same time, the lower target density and physiologic (for a T-cell) binding affinity (typically in the micromolar range for naturally occurring TCRs, compared to the nanomolar affinities of CARs [14]) facilitate a deeper penetration of TCR-T cells into solid tumors, while CAR-T cells can be halted at the outer tumor layer [10]. The downside is MHC-restriction, with most TCR-T trials focusing on HLA-A*0201-positive patients, who comprise approximately 50% of Caucasians [20].
Table 1. Comparison of chimeric antigen (CAR) and T-cell receptors (TCRs).
| CAR | TCR | |
|---|---|---|
| Target Ag | Surface proteins, glycoproteins, glycolipids, carbohydrates | Peptides from surface and intracellular proteins |
| Ag recognition | MHC-independent | MHC-dependent |
| Receptor structure | Single-chain, scFv 3 ITAMs | αβ heterodimer 10 ITAMs |
| Affinity for target | Nanomolar range | Micromolar range |
| Required target density for response | >103/cell | ∼1–50/cell |
Ag: antigen
When targeting solid tumors, the advantages of TCR over CAR are especially relevant and acquire critical importance. While several differentiation antigens expressed in the hematologic lineages are dispensable and therefore suitable for immunologic attack, for example CD19, CD33 and BCMA [21], there is no corresponding target that can be safely eliminated in epithelial cancers. TCRs directed against tumor-associated antigens (TAA) can result in serious off-tumor on-target toxicity, for example uveitis, labyrinthitis, vitiligo, and death of melanoma patients upon targeting the melanocytic differentiation antigens MART-1 and gp100, due to cross-reaction with normal melanocytes and cytokine release [22][23]. In addition, naturally occurring TCRs against TAA and cancer-testis antigens (CTA) are usually of low affinity, as TCRs with higher affinity against self-antigens are subject to negative selection in the thymus [24]. This is problematic, because lower TCR affinity has generally been linked to weaker T-cell responses in vitro and in vivo [25]. Immunization of transgenic humanized mice with human TAA to circumvent self-tolerance, amino acid substitutions, T-cell display systems and other methods are available to engineer higher-affinity TCRs [26][27][28][29][30], but also pose a significant safety risk, as illustrated by a clinical trial of a high-avidity TCR against the carcinoembryonic antigen (CEA): three patients nearly died of severe inflammatory colitis, because the transduced T cells attacked normal colonic epithelial cells, which also express CEA, albeit at lower levels [31]. In another trial using an HLA-A*0201-restricted, affinity-enhanced TCR targeting MAGE-A3/9, unexpected recognition of an HLA-A*0201-restricted MAGE-A12 epitope in the brain caused fatal neurotoxicity in two patients [32], while unexpected cross-reactivity of another affinity-enhanced TCR against HLA-A*01-restricted MAGE-A3 with the myocardial protein titin lead to cardiogenic shock and subsequent death of the first two treated patients [33]. Counterexamples of exceptional anti-tumor efficacy without off-target toxicities also exist [34][35], but testing for potential cross-reactivity is now routinely performed during the pre-clinical development of all novel TCRs [36].
Another critical problem for ACTs is the susceptibility of single-target approaches to tumor escape via antigen-loss. Clinical trials of CD19-directed CAR-T cells have indeed shown treatment failures to be mostly CD19-negative [37][38], and the same limitation also pertains to TCR-based ACT, necessitating the use of multivalent products [39]. Based on preclinical models, it has been proposed that a combination of TCRs targeting three or more mutant peptides with adequate affinity may be necessary and sufficient to eradicate established cancers [40]. Considering the scarcity, low efficiency, and poor safety of TAA and CTA, as outlined in the previous paragraph, exploitation of the much larger and much more tumor-specific reservoir of neoantigens becomes unavoidable for ACT. For the approximately 12% of human cancers attributable to oncogenic viruses, targeting oncoviral antigens would also offer similar advantages [41]. However, at the same time this also dictates the use of TCR-T over CAR-T cells, because neo- and onco-viral antigens are generally intracellular and can only be targeted by the former (Table 1) [10]. The essential tumorigenic role of many neoantigens (especially public ones, such as those generated by activating oncogene mutations) compared to TAA, that hinders antigen-loss as an evasion mechanism [14], the higher affinity of TCR directed against tumor-specific antigens (TSA) compared to TAA/CTA, and the better tumor penetration of TCR-T compared to CAR-T cells, are additional features of key importance for the treatment of solid tumors, but there are some critical bottlenecks to overcome first.
3. Critical Tasks
One first bottleneck for clinical development of such mutatome-based TCR-T therapies is currently neoantigen identification. The first step is usually whole-exome sequencing (WES) of tumor and normal tissue in order to identify non-synonymous mutations [42], followed by RNA sequencing (RNA-seq) in order to characterize the expression of altered sequences [43]. Of note, it is now possible to perform WES on cell-free tumor DNA (ctDNA) or circulating tumor-cell (CTC) DNA, which is enriched for mutations shared between primary and metastatic sites [13]. Subsequently, potential neoantigens are assessed for their capacity to be processed by the proteasome and presented on the patient’s MHC, either by bioinformatic analysis, or by mass-spectrometry-based immunopeptidomics [43][44][45][46]. Multiple studies have found that only about 1–2% of non-synonymous mutations result in neoantigens that are recognized by T cells [47]. In silico prediction of MHC-I binding for potential neoepitopes is mainly based on neural network algorithms, e.g., NetMHC, which are less accurate for infrequent HLA-I alleles, HLA-II molecules, and potential targets resulting from special alterations, e.g., long insertions/deletions, gene fusions, splicing aberrations, epigenetic changes, and posttranslational modifications [42][45]. Alternatively, peptides presented on HLA molecules can be eluted and their amino acid sequence determined using liquid-chromatography-coupled tandem MS (LC-MS/MS), which reduces the number of false positives compared to bioinformatic pipelines, and can occasionally detect cryptic peptides overlooked by in silico methods [48]. Still, while highly specific, immunopeptidomic approaches suffer from low sensitivity, especially for peptides that are less abundant and more difficult to ionize and fragment, or when the quantity of available tumor material is limited [43].
Identification of neoantigen-specific TCRs is achieved by testing the immunogenicity of potential neoepitopes against T cells [46]. Usually, these cells are collected from tumor biopsies or the patient’s blood, and can be enriched for tumor-reactive clones by sorting for CD137, CD39 and PD-1 positivity, in order to increase yield [49][50][51][52]. These can then be tested against large numbers of putative neoepitopes by high-throughput assays using barcoded pMHC-peptide multimers in microfluidic systems with a high sensitivity (detection of down to 1 in 106 neoepitope-specific cells), followed by isolation, profiling and TCR sequencing [49][53]. Suitable peptide-MHC-I multimers are meanwhile available for almost all patients (>95%), but interrogation of MHC-II remains problematic [43]. A more time-consuming T-cell screening method utilizes coculture with antigen-presenting cells (APCs) that are either transfected with “tandem minigenes” (TMGs) or pulsed with long peptides to present the patient’s neoepitopes [7][54][55]. A minigene consists of one non-synonymous mutation flanked by 12 amino acids of the wild-type sequence, and is merged together with 5–23 other similar sequences in tandem, followed by electroporation into autologous APCs, typically B cells or dendritic cells [43]. Alternatively, 25-amino-acid-long peptides encompassing the mutated amino acid can be synthesized and pulsed onto the APCs. In both cases, APCs are cocultured with either TILs or peripheral blood T cells, the reactivity of which can be assessed by ELISPOT or the upregulation of T-cell activation markers (e.g., CD137, CD134) [56][57][58]. APC-based screening is less biased than approaches relying on pMHC-multimers, and has a special advantage in the identification of neoantigen reactive CD4+ T cells. Nevertheless, costs for both methods are still high, and availability of APCs or T cells can be limiting, especially for tumors with a high tumor mutational burden (TMB) and a large number of candidate neoepitopes [43]. One solution to is to perform the screening using allogeneic T cells and APCs from healthy donors (HLA-matched or partially matched), which could recognize human tumors from different hosts in several studies, including neoantigens ignored by the patient’s own T cells [59][60][61][62]. An additional advantage of the allogeneic approach is considerable shortening of the procedure, which is essential for patients with metastatic malignancies [63]. Modern protocols can identify neoepitope-specific T cells from healthy donor T-cell repertoires in only 2 weeks [60][64][65]. On the downside, using allogeneic TCRs requires thorough evaluation for self-reactivity, as these have not been subject to selection by the patient’s thymus [40]. Currently, the fastest pipelines can complete all steps of neoantigen-specific T-cell isolation, from mutation calling to validation of immunogenicity, in 6 weeks [49].
Finally, the identified neoepitope-specific TCRs are transferred into suitable recipient cells, which are subsequently expanded to form the cellular therapeutic. Retro- and lentiviral vectors have been the mainstay for T-cell engineering for years and are still widely used. However, these procedures are associated with significant biosafety hazards that limit availability, are very expensive (approximately a quarter million USD per gene transfer), time-consuming (several weeks), and not suitable for the upscaling necessary to meet the increasing demand imposed by neoantigen multitargeting in conjunction with other modifications (Figure 1) [11][14]. Therefore, non-viral methods of targeted integration into the TCR locus have emerged as the preferred alternative, and offer the additional advantage of concomitantly disrupting the endogenous TCR, which prevents graft-versus-host disease, TCR mispairing and competition for signaling components [11]. Most promising is an entirely non-viral, CRISPR-based approach, which allows efficient, site-specific insertion of large DNA sequences (>1 kb) in the genomes of primary human T cells within 1 week, while preserving cell viability [66]. Other methods combine non-viral TCR disruption (e.g., via CRISPR, zinc-finger (ZFN) or transcription activator-like effector nucleases (TALENs)) with transduction [67][68][69][70][71], or utilize electroporation of transposons, which also reduces manufacturing costs and duration compared to viral vectors, but may impair cell viability [72] and result in a lower transgene expression [73]. Pivotal studies of CRISPR editing with ssDNA as donor template for homology-directed repair have shown a very low (0.01%) off-target integration rate [66], but genetic characterization of gene-edited cells remains essential to ensure safety [11]. Further quality measures include validation of the neoepitope specificity for TCR engineered cells and screening for off-target reactivity. Clinical trials of non-viral gene editing to simultaneously target multiple neoantigens in various cancers are currently underway (e.g., NCT04102436 and NCT03970382).
Deployment of several different neoepitope-specific TCRs for targeting multiple neoantigens in each patient classically entails manufacturing multiple mono-specific TCR-T cells, which are then pooled together or sequentially infused to the patient. However, experience with CAR-T cells has shown that co-expression of two different antigen receptors on the same T-cell results in a higher potency than pooling two different monospecific T-cell populations together [81]. The presence of naturally occurring dual-TCR T cells in the human immune system (estimated as about 10% of αβ T cells) and their enhanced alloreactivity [82,83] actually suggest that bispecific TCR-T cells might be a viable ACT option, but this remains to be explored.
Deployment of several different neoepitope-specific TCRs for targeting multiple neoantigens in each patient classically entails manufacturing multiple mono-specific TCR-T cells, which are then pooled together or sequentially infused to the patient. However, experience with CAR-T cells has shown that co-expression of two different antigen receptors on the same T-cell results in a higher potency than pooling two different monospecific T-cell populations together [74]. The presence of naturally occurring dual-TCR T cells in the human immune system (estimated as about 10% of αβ T cells) and their enhanced alloreactivity [75][76] actually suggest that bispecific TCR-T cells might be a viable ACT option, but this remains to be explored.
Current ACTs mainly depend on CD8+ T lymphocytes as cytotoxic executors [77]. However, ample evidence suggests that concomitant mobilization of CD4+ T cells against the tumor is essential for epitope spreading and the durability of CD8+ T-cell responses [78][79]. Serious obstacles to such combined strategies at present are the low efficiency of technologies for MHC-II neoepitope identification and pMHC-II-specific TCR isolation, as already outlined. In addition, most solid tumors express only pMHC-I, which natural CD4+ cells cannot recognize, while equipping them with pMHC-I-specific TCRs does not entirely solve the problem, because these generally need the participation of a CD8 co-receptor to engage the antigen [10]. One emerging solution is co-transduction with CD8α homodimers or CD8αβ, which can significantly enhance CD4+ TCR-T-cell activation and cytokine production in preclinical models [80][81]. Besides, many neoantigen-specific TCRs have a high pMHC affinity and could therefore elicit T-cell activation regardless of CD8 [82].
Beyond normal T lymphocytes, several other cell types can also be equipped with a TCR for neoantigen targeting and offer special advantages. TCR-transduced NK cells retain their natural cytotoxicity in addition to the newly-acquired MHC-restricted capabilities, and are therefore resistant to HLA-loss, which is a main mechanism of tumor evasion under TCR therapy [83][84][85]. Induced pluripotent stem cells (iPS) can be used to transform derived lymphocytes or other mesenchymal cells to cytotoxic T cells of any specificity with extended proliferative potential [86]. Gamma-delta T cells transduced with tumor-specific αβTCRs acquire MHC-restricted cytotoxic potential without the problem of TCR mispairing [87]. Of note, all aforementioned parental cell types have reduced or no alloreactivity and could also form the basis for readily available “off-the-shelf” ACTs [83][87][88].
Optimization of TCR specificity, while representing the first and crucial step, is far from the finish line in the development of an engineered T-cell therapeutic solution. Countless other interactions before and after the TCR-pMHC engagement are critical for T-cell function and need to be taken into consideration, as well. In fact, the single most important step for the breakthrough of CAR-T cells was not related to their antigen-specificity, but rather came through other improvements. Early, first-generation CARs contained only the intracellular part of the CD3ζ chain and showed little capacity to initiate an immune response in transgenic mice due to insufficient CAR-T cell activation and proliferation upon antigen engagement [83]. Only after supplementation with additional co-stimulatory receptor domains, CD28 or 4‑1BB-derived [89][90], could these second-generation CARs deliver the efficacy that ultimately led to CAR-T cell approval for hematological malignancies by the U.S. Food and Drug Administration (FDA) in 2017[91]. Improved understanding of T-cell physiology and the additional challenges posed by the microenvironment of solid tumors continuously shape further “next-generation” improvements. Many of them were first implemented in CAR-T cells, which are more advanced in development, but all are equally relevant for TCR therapies, as well. Indeed, several have already been successfully introduced into the TCR-T space, while many others are in the process of being transferred. Based on their objective, they can broadly be categorized as follows: i) improvement of T-cell persistence, memory, and fitness (e.g. engineered PD‑1 “dominant negative” receptors (DNR), or chimeric PD1/CD28 “switch” receptors [92][93]); ii) enhancement of T-cell trafficking and activation in the TME (e.g. transduction of chemokine receptors, such as CXCR2 [94]; iii) prevention of toxicity (e.g. combinatorial antigen-sensing circuits, utilizing for example the synNotch receptor [95], or suicide gene technologies, like the herpes-simplex-virus thymidine-kinase (HSV-TK) and the inducible Caspase9 system [96][97]). Some important examples are shown in Figure 1, while the entire spectrum of next-generation modifications has been extensively reviewed elsewhere [98].
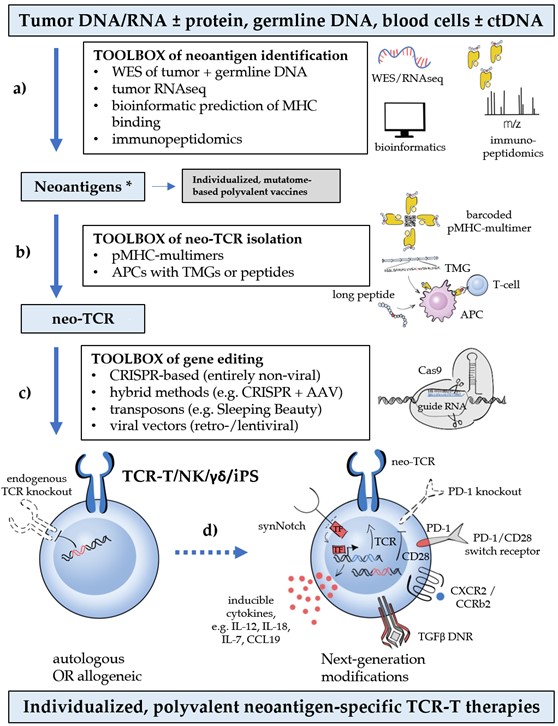
Figure 1. Critical steps, bottlenecks, and breakthroughs in neoantigen-based T-cell-receptor (TCR) therapy (from [99]). Critical steps (blue boxes), bottlenecks (shown with lower-case letters: a) rapid, high-throughput identification of public and private neoantigens; b) isolation of neoepitope-specific TCRs (neo-TCRs); c) (preferably non-viral) gene editing of autologous or allogeneic cells with concomitant knock-out of the endogenous TCR; d) additional next-generation modifications to improve T-cell physiology), and technological breakthroughs (white boxes) that drive progress in the TCR therapy of cancer. The term “third-generation ACTs” has been coined for products combining these new technologies. Polyvalency currently entails manufacturing multiple mono-specific TCR-T cells, which are then pooled together or sequentially infused to the patient. * in case of virally induced tumors, oncoviral antigens are also tumor-specific and can be exploited similarly to the tumor neoantigens.
4. Status of Clinical Development
The growing interest for cancer TCR-T therapies and the increasing number of promising strategies are reflected in the upsurge of publications and clinical trials during recent years (Figure 2a). The first oncological studies with engineered TCR-T cells were registered at clinicaltrials.gov as early as 2004, and with some fluctuations, their numbers have increased markedly over the last 15 years (Figure 2).
All registered studies are either phase I or II, and the vast majority concern solid tumors rather than hematological malignancies (Figure 2b). The most prevalent entity is melanoma, followed by gastrointestinal cancers, lung cancer, and almost all other solid tumors (Figure 2c). Most frequent targets are CTA and other TAA, while oncoviral antigens and neoantigens make up < 20% each (Figure 2b). Targeting of multiple neoantigens is even less frequent (<10%, Figure 2b), with the earliest such trial registered only in 2018 (NCT03412877). Non-viral gene editing as implemented within the NCT04102436 trial is the newest development to enter clinical testing, with an estimated start date in Q4 2020.
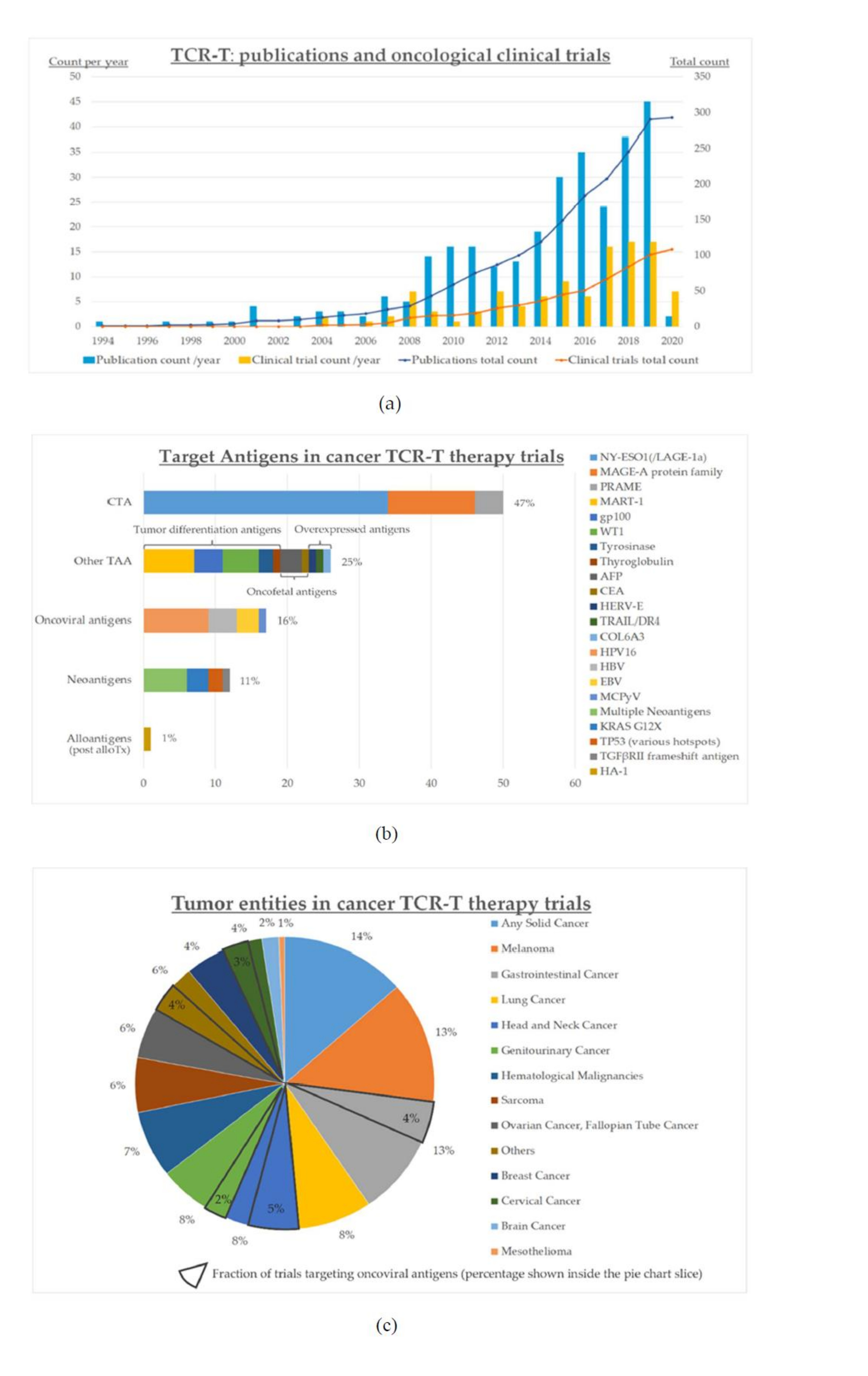
Figure 2. Status quo of clinical development for cancer TCR-T therapies as of June 2020: (a) numbers of clinical trials (n=104) and publications (n=293); (b) target antigens in the various clinical trials; (c) cancer entities in the various clinical trials; “others” includes vulvar (n=3) and vaginal (n=2) neoplasms, primary peritoneal carcinoma (n=2), thyroid cancer (n=1), and Merkel-cell carcinoma (n=1). Clinical trials were identified by a search in ClinicalTrials.gov on June 15, 2020 using the keyword “TCR”, followed by filtering the results to include interventional trials for oncological entities only, and manually verifying which trials specifically employ genetically engineered TCR-T therapies (n=104). Publications were identified by a search in PubMed using ((“Immunotherapy, Adoptive”[Mesh]) AND (TCR[Title/Abstract])) OR ((“Immunotherapy, Adoptive”[Mesh]) AND (T cell receptor[Title/Abstract])), which returned 853 entries, followed by manual verification of TCR-T therapies as the main subject (n=293, publications on other ACT, e.g., CAR-T, and studies not involving TCR engineering, e.g., using transgenic mouse models, were excluded); alloTx: allogeneic hematopoietic cell transplantation.
5. Conclusions and Perspectives
The field of oncological TCR therapies is extremely versatile and evolves rapidly. Currently, two elements emerge as the most promising aspects for therapeutic potential and future clinical impact: individualized, mutatome-based strategies utilizing high-throughput neoantigen-specific TCR-isolation and expeditious non-viral gene editing; as well as “next-generation” modifications that improve T‑cell physiology and boost immune activation in the adverse solid tumor microenvironment. Facilitated by major technological breakthroughs, both can meanwhile be realized within clinical trials and herald the next immuno-oncological revolution.
This entry is adapted from the peer-reviewed paper 10.3390/cells9092095
References
- Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013, 342, 1432–1433.
- Garon, E.B.; Hellmann, M.D.; Rizvi, N.A.; Carcereny, E.; Leighl, N.B.; Ahn, M.-J.; Eder, J.P.; Balmanoukian, A.S.; Aggarwal, C.; Horn, L.; et al. Five-year overall survival for patients with advanced non‒small-cell lung cancer treated with pembrolizumab: Results from the phase I KEYNOTE-001 study. J. Clin. Oncol. 2019, 37, 2518–2527.
- Gettinger, S.; Horn, L.; Jackman, D.; Spigel, D.; Antonia, S.; Hellmann, M.; Powderly, J.; Heist, R.; Sequist, L.V.; Smith, D.C.; et al. Five-year follow-up of nivolumab in previously treated advanced non-small-cell lung cancer: Results from the CA209-003 study. J. Clin. Oncol. 2018, 36, 1675–1684.
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2019, 381, 1535–1546.
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers 2020, 12, 738.
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018, 8, 1069–1086.
- Xin Yu, J.; Hubbard-Lucey, V.M.; Tang, J. Immuno-oncology drug development goes global. Nat. Rev. Drug Discov. 2019, 18, 899–900.
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.-H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684.
- Lee, Y.-H.; Kim, C.H. Evolution of chimeric antigen receptor (CAR) T cell therapy: Current status and future perspectives. Arch. Pharmacal Res. 2019, 42, 607–616.
- Jafarzadeh, L.; Masoumi, E.; Fallah-Mehrjardi, K.; Mirzaei, H.R.; Hadjati, J. Prolonged persistence of Chimeric Antigen Receptor (CAR) T cell in adoptive cancer immunotherapy: Challenges and ways forward. Front. Immunol. 2020, 11, 702.
- Berger, C.; Jensen, M.C.; Lansdorp, P.M.; Gough, M.; Elliott, C.; Riddell, S.R. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J. Clin. Investig. 2008, 118, 294–305.
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419.
- Klebanoff, C.A.; Rosenberg, S.A.; Restifo, N.P. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat. Med. 2016, 22, 26–36.
- Chandran, S.S.; Klebanoff, C.A. T cell receptor-based cancer immunotherapy: Emerging efficacy and pathways of resistance. Immunol. Rev. 2019, 290, 127–147.
- Harris, D.T.; Kranz, D.M. Adoptive T cell therapies: A comparison of T cell receptors and chimeric antigen receptors. Trends Pharmacol. Sci. 2016, 37, 220–230.
- Zhao, L.; Cao, Y.J. Engineered T cell therapy for cancer in the clinic. Front. Immunol. 2019, 10, 2250.
- Valitutti, S.; Müller, S.; Cella, M.; Padovan, E.; Lanzavecchia, A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature 1995, 375, 148–151.
- Sykulev, Y.; Joo, M.; Vturina, I.; Tsomides, T.J.; Eisen, H.N. Evidence that a single peptide–MHC complex on a target cell can elicit a cytolytic T cell response. Immunity 1996, 4, 565–571.
- Stauss, H.J. Turn to TCRs when CARs fail. Oncotarget 2017, 8, 12538–12539.
- Ellis, J.M.; Henson, V.; Slack, R.; Ng, J.; Hartzman, R.J.; Katovich, H.C. Frequencies of HLA-A2 alleles in five U.S. population groups. Hum. Immunol. 2000, 61, 334–340.
- Rotolo, A.; Karadimitris, A.; Ruella, M. Building upon the success of CART19: Chimeric antigen receptor T cells for hematologic malignancies. Leuk. Lymphoma 2018, 59, 2040–2055.
- Johnson, L.A.; Morgan, R.A.; Dudley, M.E.; Cassard, L.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Sherry, R.M.; Wunderlich, J.R.; et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009, 114, 535–546.
- Van den Berg, J.H.; Gomez-Eerland, R.; van de Wiel, B.; Hulshoff, L.; van den Broek, D.; Bins, A.; Tan, H.L.; Harper, J.V.; Hassan, N.J.; Jakobsen, B.K.; et al. Case report of a fatal serious adverse event upon administration of T cells transduced with a MART-1-specific T-cell receptor. Mol. Ther. 2015, 23, 1541–1550.
- Gotter, J.; Brors, B.; Hergenhahn, M.; Kyewski, B. Medullary epithelial cells of the human thymus express a highly diverse selection of tissue-specific genes colocalized in chromosomal clusters. J. Exp. Med. 2004, 199, 155–166.
- Zhong, S.; Malecek, K.; Johnson, L.A.; Yu, Z.; de Vega-Saenz, M.E.; Darvishian, F.; McGary, K.; Huang, K.; Boyer, J.; Corse, E.; et al. T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proc. Natl. Acad. Sci. USA 2013, 110, 6973–6978.
- Schmitt, T.M.; Aggen, D.H.; Stromnes, I.M.; Dossett, M.L.; Richman, S.A.; Kranz, D.M.; Greenberg, P.D. Enhanced-affinity murine T-cell receptors for tumor/self-antigens can be safe in gene therapy despite surpassing the threshold for thymic selection. Blood 2013, 122, 348–356.
- Obenaus, M.; Leitão, C.; Leisegang, M.; Chen, X.; Gavvovidis, I.; van der Bruggen, P.; Uckert, W.; Schendel, D.J.; Blankenstein, T. Identification of human T-cell receptors with optimal affinity to cancer antigens using antigen-negative humanized mice. Nat. Biotechnol. 2015, 33, 402–407.
- Li, Y.; Moysey, R.; Molloy, P.E.; Vuidepot, A.-L.; Mahon, T.; Baston, E.; Dunn, S.; Liddy, N.; Jacob, J.; Jakobsen, B.K.; et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat. Biotechnol. 2005, 23, 349–354.
- Robbins, P.F.; Li, Y.F.; El-Gamil, M.; Zhao, Y.; Wargo, J.A.; Zheng, Z.; Xu, H.; Morgan, R.A.; Feldman, S.A.; Johnson, L.A.; et al. Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J. Immunol. 2008, 180, 6116–6131.
- Chervin, A.S.; Aggen, D.H.; Raseman, J.M.; Kranz, D.M. Engineering higher affinity T cell receptors using a T cell display system. J. Immunol. Methods 2008, 339, 175–184.
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.-A.N.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 2011, 19, 620–626.
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013, 36, 133–151.
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013, 122, 863–871.
- D’Angelo, S.P.; Melchiori, L.; Merchant, M.S.; Bernstein, D.; Glod, J.; Kaplan, R.; Grupp, S.; Tap, W.D.; Chagin, K.; Binder, G.K.; et al. Antitumor activity associated with prolonged persistence of adoptively transferred NY-ESO-1 c259T cells in synovial sarcoma. Cancer Discov. 2018, 8, 944–957.
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J.; et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015, 21, 914–921.
- Border, E.C.; Sanderson, J.P.; Weissensteiner, T.; Gerry, A.B.; Pumphrey, N.J. Affinity-enhanced T-cell receptors for adoptive T-cell therapy targeting MAGE-A10: Strategy for selection of an optimal candidate. Oncoimmunology 2019, 8, e1532759.
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517.
- Majzner, R.G.; Mackall, C.L. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018, 8, 1219–1226.
- Kaluza, K.M.; Kottke, T.; Diaz, R.M.; Rommelfanger, D.; Thompson, J.; Vile, R. Adoptive transfer of cytotoxic T lymphocytes targeting two different antigens limits antigen loss and tumor escape. Hum. Gene Ther. 2012, 23, 1054–1064.
- Blankenstein, T.; Leisegang, M.; Uckert, W.; Schreiber, H. Targeting cancer-specific mutations by T cell receptor gene therapy. Curr. Opin. Immunol. 2015, 33, 112–119.
- Schiller, J.T.; Lowy, D.R. Vaccines to prevent infections by oncoviruses. Annu. Rev. Microbiol. 2010, 64, 23–41.
- Anonymous. The problem with neoantigen prediction. Nat. Biotechnol. 2017, 35, 97.
- Garcia-Garijo, A.; Fajardo, C.A.; Gros, A. Determinants for neoantigen identification. Front. Immunol. 2019, 10, 1392.
- Richters, M.M.; Xia, H.; Campbell, K.M.; Gillanders, W.E.; Griffith, O.L.; Griffith, M. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med. 2019, 11, 56.
- Yarchoan, M.; Johnson, B.A.; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222.
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200.
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74.
- Purcell, A.W.; Ramarathinam, S.H.; Ternette, N. Mass spectrometry-based identification of MHC-bound peptides for immunopeptidomics. Nat. Protoc. 2019, 14, 1687–1707.
- Arnaud, M.; Duchamp, M.; Bobisse, S.; Renaud, P.; Coukos, G.; Harari, A. Biotechnologies to tackle the challenge of neoantigen identification. Curr. Opin. Biotechnol. 2020, 65, 52–59.
- Gros, A.; Robbins, P.F.; Yao, X.; Li, Y.F.; Turcotte, S.; Tran, E.; Wunderlich, J.R.; Mixon, A.; Farid, S.; Dudley, M.E.; et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J. Clin. Investig. 2014, 124, 2246–2259.
- Duhen, T.; Duhen, R.; Montler, R.; Moses, J.; Moudgil, T.; de Miranda, N.F.; Goodall, C.P.; Blair, T.C.; Fox, B.A.; McDermott, J.E.; et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun. 2018, 9, 2724.
- Wolfl, M.; Kuball, J.; Ho, W.Y.; Nguyen, H.; Manley, T.J.; Bleakley, M.; Greenberg, P.D. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood 2007, 110, 201–210.
- Puig-Saus, C.; Sennino, B.; Purandare, B.; An, D.; Quach, B.; Peng, S.; Xia, H.; Zhao, S.; Pan, Z.; Ma, Y.; et al. Landscape analysis of neoepitope-specific T-cell responses to immunotherapy. In The 111th Annual Meeting of the American Association for Cancer Research, Proceedings of the AACR Annual Meeting 2020, Philadelphia, PA, USA, 22–24 June 2020; AACR: Philadelphia, PA, USA, 2020.
- Parkhurst, M.; Gros, A.; Pasetto, A.; Prickett, T.; Crystal, J.S.; Robbins, P.; Rosenberg, S.A. Isolation of T-cell receptors specifically reactive with mutated tumor-associated antigens from tumor-infiltrating lymphocytes based on CD137 expression. Clin. Cancer Res. 2017, 23, 2491–2505.
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.-C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645.
- Tran, E.; Robbins, P.F.; Rosenberg, S.A. ‘Final common pathway’ of human cancer immunotherapy: Targeting random somatic mutations. Nat. Immunol. 2017, 18, 255–262.
- Seliktar-Ofir, S.; Merhavi-Shoham, E.; Itzhaki, O.; Yunger, S.; Markel, G.; Schachter, J.; Besser, M.J. Selection of shared and neoantigen-reactive T cells for adoptive cell therapy based on CD137 separation. Front. Immunol. 2017, 8, 1211.
- Yossef, R.; Tran, E.; Deniger, D.C.; Gros, A.; Pasetto, A.; Parkhurst, M.R.; Gartner, J.J.; Prickett, T.D.; Cafri, G.; Robbins, P.F.; et al. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight 2018, 3, e112467.
- Liu, S.; Matsuzaki, J.; Wei, L.; Tsuji, T.; Battaglia, S.; Hu, Q.; Cortes, E.; Wong, L.; Yan, L.; Long, M.; et al. Efficient identification of neoantigen-specific T-cell responses in advanced human ovarian cancer. J. Immunother. Cancer 2019, 7, 156.
- Ali, M.; Foldvari, Z.; Giannakopoulou, E.; Böschen, M.-L.; Strønen, E.; Yang, W.; Toebes, M.; Schubert, B.; Kohlbacher, O.; Schumacher, T.N.; et al. Induction of neoantigen-reactive T cells from healthy donors. Nat. Protoc. 2019, 14, 1926–1943.
- Scheper, W.; Kelderman, S.; Fanchi, L.F.; Linnemann, C.; Bendle, G.; de Rooij, M.A.J.; Hirt, C.; Mezzadra, R.; Slagter, M.; Dijkstra, K.; et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat. Med. 2019, 25, 89–94.
- Strønen, E.; Toebes, M.; Kelderman, S.; van Buuren, M.M.; Yang, W.; van Rooij, N.; Donia, M.; Böschen, M.-L.; Lund-Johansen, F.; Olweus, J.; et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 2016, 352, 1337–1341.
- Dwarshuis, N.J.; Parratt, K.; Santiago-Miranda, A.; Roy, K. Cells as advanced therapeutics: State-of-the-art, challenges, and opportunities in large scale biomanufacturing of high-quality cells for adoptive immunotherapies. Adv. Drug Deliv. Rev. 2017, 114, 222–239.
- Anczurowski, M.; Hirano, N. Two weeks’ notice from allogeneic sources. Clin. Cancer Res. 2018, 24, 5195–5197.
- Matsuda, T.; Leisegang, M.; Park, J.-H.; Ren, L.; Kato, T.; Ikeda, Y.; Harada, M.; Kiyotani, K.; Lengyel, E.; Fleming, G.F.; et al. Induction of neoantigen-specific cytotoxic T cells and construction of T-cell receptor-engineered T cells for ovarian cancer. Clin. Cancer Res. 2018, 24, 5357–5367.
- Roth, T.L.; Puig-Saus, C.; Yu, R.; Shifrut, E.; Carnevale, J.; Li, P.J.; Hiatt, J.; Saco, J.; Krystofinski, P.; Li, H.; et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 2018, 559, 405–409.
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367.
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117.
- Albers, J.J.; Ammon, T.; Gosmann, D.; Audehm, S.; Thoene, S.; Winter, C.; Secci, R.; Wolf, A.; Stelzl, A.; Steiger, K.; et al. Gene editing enables T-cell engineering to redirect antigen specificity for potent tumor rejection. Life Sci. Alliance 2019, 2.
- Provasi, E.; Genovese, P.; Lombardo, A.; Magnani, Z.; Liu, P.-Q.; Reik, A.; Chu, V.; Paschon, D.E.; Zhang, L.; Kuball, J.; et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat. Med. 2012, 18, 807–815.
- Berdien, B.; Mock, U.; Atanackovic, D.; Fehse, B. TALEN-mediated editing of endogenous T-cell receptors facilitates efficient reprogramming of T lymphocytes by lentiviral gene transfer. Gene Ther. 2014, 21, 539–548.
- Peng, P.D.; Cohen, C.J.; Yang, S.; Hsu, C.; Jones, S.; Zhao, Y.; Zheng, Z.; Rosenberg, S.A.; Morgan, R.A. Efficient nonviral sleeping beauty transposon-based TCR gene transfer to peripheral blood lymphocytes confers antigen-specific antitumor reactivity. Gene Ther. 2009, 16, 1042–1049.
- Bailey, S.R.; Maus, M.V. Gene editing for immune cell therapies. Nat. Biotechnol. 2019, 37, 1425–1434.
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.H.; et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052.
- Schuldt, N.J.; Binstadt, B.A. Dual TCR T cells: Identity crisis or multitaskers? J. Immunol. 2019, 202, 637–644.
- Balakrishnan, A.; Morris, G.P. The highly alloreactive nature of dual TCR T cells. Curr. Opin. Organ. Transpl. 2016, 21, 22–28.
- Durgeau, A.; Virk, Y.; Corgnac, S.; Mami-Chouaib, F. Recent advances in targeting CD8 T-cell immunity for more effective cancer immunotherapy. Front. Immunol. 2018, 9, 14.
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the role of CD4+ T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther. 2020.
- Sillito, F.; Holler, A.; Stauss, H.J. Engineering CD4+ T cells to enhance cancer immunity. Cells 2020, 9, 1721.
- Anderson, V.E.; Weber, A.M.; Wiedermann, G.E.; Pachnio, A.; Dauleh, S.; Ahmed, T.; Docta, R.Y.; Quattrini, A.; Pope, G.; Quinn, L.; et al. Abstract 2313: Enhanced activity of second-generation MAGE-A4 SPEAR T-cells through co-expression of a CD8α homodimer. In Immunology, Proceedings of the AACR Annual Meeting 2019, Atlanta, GA, USA, 29 March–3 April 2019; AACR: Philadelphia, PA, USA, 2019; p. 2313.
- Ghorashian, S.; Veliça, P.; Chua, I.; McNicol, A.-M.; Carpenter, B.; Holler, A.; Nicholson, E.; Ahmadi, M.; Zech, M.; Xue, S.-A.; et al. CD8 T cell tolerance to a tumor-associated self-antigen is reversed by CD4 T cells engineered to express the same T cell receptor. J. Immunol. 2015, 194, 1080–1089.
- Holler, P.D.; Kranz, D.M. Quantitative analysis of the contribution of TCR/pepMHC affinity and CD8 to T cell activation. Immunity 2003, 18, 255–264.
- Mensali, N.; Dillard, P.; Hebeisen, M.; Lorenz, S.; Theodossiou, T.; Myhre, M.R.; Fåne, A.; Gaudernack, G.; Kvalheim, G.; Myklebust, J.H.; et al. NK cells specifically TCR-dressed to kill cancer cells. EBioMedicine 2019, 40, 106–117.
- Parlar, A.; Sayitoglu, E.C.; Ozkazanc, D.; Georgoudaki, A.-M.; Pamukcu, C.; Aras, M.; Josey, B.J.; Chrobok, M.; Branecki, S.; Zahedimaram, P.; et al. Engineering antigen-specific NK cell lines against the melanoma-associated antigen tyrosinase via TCR gene transfer. Eur. J. Immunol. 2019, 49, 1278–1290.
- Anonymous. HLA loss facilitates immune escape. Cancer Discov. 2018, 8, 8.
- Minagawa, A.; Yoshikawa, T.; Yasukawa, M.; Hotta, A.; Kunitomo, M.; Iriguchi, S.; Takiguchi, M.; Kassai, Y.; Imai, E.; Yasui, Y.; et al. Enhancing T cell receptor stability in rejuvenated iPSC-derived T cells improves their use in cancer immunotherapy. Cell Stem Cell 2018, 23, 850–858.e4.
- Van der Veken, L.T.; Hagedoorn, R.S.; van Loenen, M.M.; Willemze, R.; Falkenburg, J.H.F.; Heemskerk, M.H.M. Alphabeta T-cell receptor engineered gammadelta T cells mediate effective antileukemic reactivity. Cancer Res. 2006, 66, 3331–3337.
- Iriguchi, S.; Kaneko, S. Toward the development of true “off-the-shelf” synthetic T-cell immunotherapy. Cancer Sci. 2019, 110, 16–22.