Biofunctional peptide FNIII14, which is derived from the 14th fibronectin (FN) type III-like (FN-III) repeat of FN molecule, is capable of inhibiting cell adhesion to the extracellular matrix (ECM). This functional site is usually buried within the molecular structure of FN, but can be exposed by conformational changes and proteolytic cleavage. Peptide FNIII14 can induce a conformational change in β1-integrin from the active to the inactive form, causing functional inactivation. Based on this anti-adhesive activity, peptide FNIII14 exhibits therapeutic potential for several diseases such as metabolic diseases, organ fibrosis, and malignant tumors. Peptide FNIII14 blocks integrin-mediated signaling by a mechanism entirely distinct from that of conventional antagonisitic peptides, including Arg-Gly-Asp peptides that competitively inhibit the ECM binding of integrin.
- extracellular matrix
- fibronectin
- FNIII14
- integrin inactivation
- cell adhesion
- anti-fibrotic activ-ity
- anti-cancer activity
1. Introduction
Extracellular matrix (ECM) protein molecules harbor functional sites within their molecular structures [1]. Some, such as sites for cell adhesion, including integrin-binding sites represented by the Arg-Gly-Asp (RGD) motif of fibronectin (FN) and vitronectin, are exposed on the surface of protein molecules [2]. In contrast, other functional sites are often buried within the molecular structure of ECM proteins [3]. These cryptic functional sites, referred to as matricryptic sites, are exposed through proteolytic cleavage by proteinases during tissue remodeling. They also become functional by structural unfolding through cell adhesion and interaction with other ECM components in response to temporal and spatial changes in the microenvironment [3]. Peptides/fragments containing these matricryptic functional sites have unique biological activities not often detected in the parent molecules [1,3,4].
FN, an abundant and ubiquitous ECM protein, is an essential cell adhesion protein, and has several integrin-binding sites, which provide a scaffold for adhesion to various types of cells. Two types of FN are present in vertebrates: soluble FN produced by hepatocytes secreted into plasma (plasma FN), and an insoluble form of FN, which is the major component of the ECM (cellular FN) [5]. Both FN molecules not only play important roles in wound healing as cell adhesion substrates but can also regulate fundamental cellular functions, including cell survival, proliferation, differentiation, and migration. Several adhesion sequences within the FN molecule have been intensively studied [6,7], including the sequences Leu–Asp–Val (LDV) and Arg-Glu-Asp-Val (REDV) in the type III connecting segment (IIICS) domain, which are recognized by integrin α4β1, as well as the sequence Pro-His-Ser-Arg-Asn (PHSRN) and RGD in the 9th and 10th type III repeat, respectively, both of which are recognized by integrin α5β1. FN contains fibrin-, heparin-, collagen-, and cell-binding domains, each of which comprises type I, II, and III repeats (Figure 1a). Given its versatile domains and structure, FN plays a central role in ECM–cell interactions and mediates flexible signaling via these interactions.
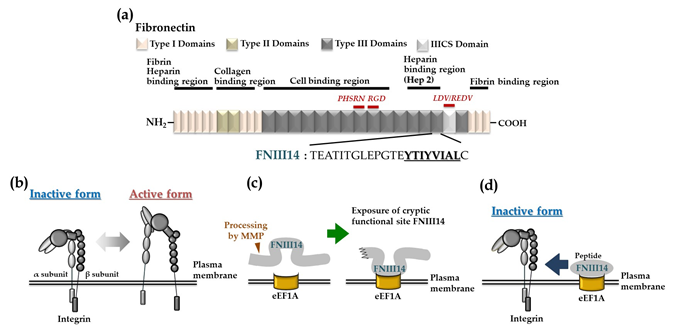
Figure 1. The fibronectin-derived functional peptide FNIII14. (a) Schematic representation of human fibronectin and the amino acid sequence of the anti-adhesion peptide FNIII14. (b) Conformational regulation of integrin structure. (c) Exposure of the cryptic anti-adhesive site of FNIII14 within the FN molecule by matrix metalloproteinase (MMP) processing. (d) Peptide FNIII14 induces a conformational change in integrin from the active to an inactive state via its putative membrane receptor eukaryotic elongation factor 1A (eEF1A). Figures modified and revised from [8].
However, the FN molecule is susceptible to proteolytic degradation by inflammatory proteinases, such as matrix metalloproteinases (MMPs) and thrombin, exposing the matricryptic sites as bioactive peptides exhibiting a variety of biological functions [1,4,8]. We found that FN harbors a matricryptic site with the amino acid sequence YTIYVIAL within the 14th FN type III domain. This site inhibits cell adhesion to the ECM. This cryptic anti-adhesion site is exposed by the interaction of the carboxyl-terminal heparin-binding domain (Hep 2) with ECM components containing sulfated sugars such as heparin. Peptide FNIII14, a 22-mer FN peptide containing this site, can induce conformational change in β1-integrin from the active to the inactive form, resulting in functional inactivation.
2. Peptide FNIII14 Derived from the FN Molecule
2.1. Anti-Adhesion Site of Fibronectin and Its Pepide FNIII14
We found that an FN fragment with a molecular mass of 30 kDa and containing the Hep 2 domain is generated through proteolytic degradation of the FN molecule. This fragment inhibits integrin-mediated cell adhesion and cell migration, which are the opposite effects of intact FN [9]. The fragment exhibits this anti-adhesive effect without binding to the intact FN molecule, indicating that the anti-adhesive effect is not the result of it masking the adhesion sites of the FN molecule [10]. In addition, the FN fragment shows anti-adhesive activity when coated on a culture dish as an insoluble substrate, as well as when added as a soluble factor, suggesting that the anti-adhesive effect may be expressed via a putative cell surface receptor [10]. Flow cytometric analysis showed the presence of a specific binding molecule for the FN fragment on the cell surface [10]. These results indicate that FN contains a cryptic molecular region with anti-adhesive activity that is probably transduced by a putative cell surface receptor [10].
This anti-adhesive effect disappeared by repeated freezing and thawing of the FN fragment solution but was restored by treatment with a denaturant such as high-concentration urea, indicating that the anti-adhesive site is buried within the molecular structure of the Hep 2 domain and the native FN protein molecule. Analysis of light scattering in circular dichroism spectra showed that urea treatment induced a conformational change in the FN fragment, from a more folded form to an unfolded form [11]. Moreover, incubation of the FN fragment with heparin induced conformational changes similar to those treated with urea, leading to the expression of anti-adhesive activity [11]. Limited proteolytic cleavage of either the FN fragment or native FN molecule by MMP-2 released a fragment with cryptic anti-adhesive activity [11]. Thus, it was found that the FN molecule harbors a cryptic anti-adhesion site within its molecular structure, which becomes bioactive through molecular processes such as conformational change and proteolytic cleavage [11].
We determined the amino acid sequence of the anti-adhesion site by characterizing the abilities of several synthetic peptides corresponding to segments of the primary structure of the FN fragment containing the Hep 2 domain to decrease the adhesion of A375SM human melanoma cells to FN substrate [12]. One peptide (TEATITGLEPGTEYTIYVIAL, residues 1835-1855 in the 14th FN type III domains, Figure 1a) remarkably suppressed cell adhesion to FN substrate. The anti-adhesive activities of sub-peptides and scrambled peptides showed that the hydrophobic moiety YTIYVIAL (residues 1848–1855) is indispensable for this activity [12]. A 22-mer synthetic peptide containing YTIYVIAL, called peptide FNIII14, induced a conformational change in β1-integrin from the active to an inactive form, as judged by flow cytometry analysis using a monoclonal antibody that recognizes an active conformation-specific epitope of β1-integrins (Figure 1b) [12]. These results suggest that anti-adhesive activity is closely associated with the sequence YTIVIAL, typically buried within the structure of 14th FN type III structure due to its hydrophobic nature [12].
125I-labeled FN fragment containing the RGD cell adhesion site binds to the cell surface via β1-integrin and was dissociated by peptide FNIII14, with concomitant binding of peptide FNIII14 to the cell surface, suggesting that peptide FNIII14 inactivates β1-integrin through binding to a membrane receptor [13]. Binding analysis by affinity labeling using biotinylated peptide FNIII14 detected a non-integrin membrane protein with a molecular mass of around 50 kDa (“p50”) as a putative membrane receptor with specific binding activity to peptide FNIII14 [14]. Surprisingly, the N-terminal amino acid sequence of p50 was identical to that of eukaryotic elongation factor 1A (eEF1A) [14]. Indeed, immunofluorescence analysis using a monoclonal antibody directed to eEF1A demonstrated the presence of eEF1A on the outer surface of cells. In addition, the anti-adhesive effect of peptide FNIII14 was correlated with the expression level of eEF1A on the cell membrane, suggesting that p50 acts as a membrane receptor mediating the anti-adhesive effect of peptide FNIII14 [14]. eEF1A is known to play a key canonical role during protein biosynthesis by ribosomes. Our findings indicate that a minor part of eEF1A is localized on the cell surface and that it similarly acts as a membrane receptor mediating the anti-adhesive effect of peptide FNIII14. We propose that peptide FNIII14 stabilizes an inactive conformation of β1-integrin by inducing lateral binding of the membrane eEF1A and β1-integrin, thereby exhibiting the anti-adhesive effect (Figure 1c, d). It has been reported that cell adhesion via integrins is involved not only in many physiological processes but also in various pathological phenomena. Below we show the possibility of suppressing some pathological processes by using the anti-adhesive effect of peptide FNIII14 by inactivating β1-integrin.
2.2. Involvement of Integrin-Mediated Adhesion to the ECM in Adipocyte Differentiaion and Related Metabolic Disorder
Cell differentiation is often accompanied by alterations in cell morphology that are highly dependent on adhesive interaction with the ECM architecture. The expression levels of factors determining cell morphology, including FN, integrins, and several cytoskeletal proteins, are reported to be changed during adipogenesis [15–17], and thus differentiation of preadipocytes into adipocytes may also be influenced by ECM-to-cell adhesion. We found that the exogenous addition of FN to an in vitro culture of the mouse preadipocyte cell line ST-13 almost completely inhibited insulin-induced adipocyte differentiation [13]. FN fibrillogenesis around the ST-13 cells was highly enhanced, and the cells showed a well-extended and flattened morphology [13]. Peptide FNIII14 accelerated the insulin-induced adipocyte differentiation of ST-13, likely by weakening ST-13 cell adhesion to the FN substrate by inactivating β1-integrin and suppressing FN fibrillogenesis [13]. In support of our observations, it was previously reported that surface expression of a disintegrin and metalloprotease 12 inactivates the function of β1-integrins and alters the organization of the actin cytoskeleton and FN-rich ECM during an early phase of adipocyte differentiation [17]. Taking all these results together, it can be concluded that peptide FNIII14 promotes adipocyte differentiation of ST-13 cells by inactivating β1-integrin. Accordingly, β1-integrins may provide a strategy for controlling obesity and obesity-associated metabolic diseases, including insulin resistance.
2.3. Anti-Fibrotic Activity of Peptide FNIII14
Myofibroblastic conversion of hepatic stellate cells is considered central to the pathogenesis of liver fibrosis. Hepatic stellate cells are usually quiescent, with low proliferative potential, and act as vitamin A storage cells, but are converted to myofibroblasts by inflammatory stimulation and become proliferative and capable of forming fibers. The inactivation of β1-integrin by peptide FNIII14 was shown to inhibit the myofibroblastic conversion of rat hepatic stellate cells (HSC) [18]. Freshly isolated rat HSC differentiate into myofibroblasts during culture with fetal bovine serum, as determined by various phenotypic activation characteristics of rat HSC such as increased proliferation, loss of vitamin A-rich lipid droplets, and expression of α-smooth muscle actin, but these characteristics are impeded by peptide FNIII14 [18]. Moreover, activated rat HSC acquire the myofibroblastic phenotype through repeated subculture, secrete FN, and stimulate the matrix assembly of FN, but all these features are inhibited by suppressing the adhesive interaction of rat HSC using peptide FNIII14 [18]. Taken together, peptide FNIII14 might suppress the myofibroblastic conversion of rat HSC by inhibiting the integrin-mediated adhesive interaction of rat HSC [18]. In support of our observations, Martin and colleagues demonstrated that β1-integrin expression levels are increased in activated rat HSC compared with quiescent rat HSC. Furthermore, they showed that the inhibition of β1-integrin signaling could suppress pro-fibrotic features of activated HSC in vitro, including expression of α-smooth muscle actin, cell migration, proliferation, contraction, and collagen deposition [19]. These results support our proposal that inactivation of β1-integrin could be a strategy for anti-liver fibrosis therapy, and peptide FNIII14 may provide an effective tool for β1-integrin inactivation.
2.4. Anti-Cancer Effects of Peptide FNIII14
Inactivation of β1-integrins by peptide FNIII14 exerts potent anti-cancer effects on several solid tumors and hematologic malignancies. We and others have demonstrated the anti-cancer effects of peptide FNIII14 on several malignant tumors both in vitro and in vivo (Table 1). For example, glioblastoma develop their characteristic aggressive properties, such as hyperproliferation and disseminative migration, through potent and sustained activation of β1-integrin, and peptide FNIII14 can inhibit the development of these aggressive properties [20]. In addition, peptide FNIII14 inhibits the anchorage-independent survival of glioblastoma cells and overcomes their anoikis resistance [21]. In vivo experiments using a mouse xenograft model demonstrated that peptide FNIII14 also suppresses tumor growth [20]. Furthermore, many clinical reports have shown that glioblastoma exhibit therapeutic resistance to temozolomide, a first-line chemotherapeutic alkylating agent for glioblastoma therapy, due, at least in part, to DNA repair by O6-methylguanine-DNA methyltransferase (MGMT) [20]. It is noteworthy that peptide FNIII14 sensitized glioblastoma cells to temozolomide by down-regulating MGMT. In addition, peptide FNIII14 was shown to augment temozolomide-induced anti-cancer efficiency in a subcutaneous mouse xenograft model [20]. β1-integrins, the target of peptide FNIII14, are highly expressed in glioblastoma, especially mesenchymal glioblastoma, which is the most aggressive phenotype of glioblastoma, and its expression levels are correlated with poor prognosis in high-grade glioma [22,23]. The β1-integrin signaling pathway confers resistance to temozolomide, radiation therapy, and anti-angiogenic therapy with bevacizumab [22,24–26]. Inhibition of β1-integrins has been shown to attenuate tumor growth and to augment anti-angiogenic therapy in a glioblastoma xenograft model [26]. Taken altogether, the application of peptide FNIII14 as a monotherapy or in combination with alkylating agents may result in a new perspective on the treatment of glioblastoma patients with poor prognosis for long-term survival.
Based on β1-integrin inactivation, peptide FNIII14 also impeded the development of other malignant properties such as anchorage-independent survival/proliferation and invasive migration in neuroblastoma cells with MYCN amplification, which are the most reliable risk factors in neuroblastoma patients [27]. Peptide FNIII14 can induce a remarkable decrease in N-Myc protein levels in neuroblastoma cells due to proteasomal degradation [27]. Peptide FNIII14 monotreatment can inhibit tumor growth in a mouse xenograft model [27]. Furthermore, proteasomal degradation caused by the conformational inactivation of β1-integrin by peptide FNIII14 is observed with another Myc family oncoprotein, c-myc, leading to abrogation of its malignant properties in pancreatic cancer cells [27]. Since Myc family oncoproteins are highly expressed in various cancers and are involved in the expression of various malignant properties, the clinical use of peptide FNIII14 is highly promising.
Peptide FNIII14 also has an anti-metastatic activity. When murine T lymphoma cells were injected into the tail vein of mice, metastases to the liver and spleen caused the weight of these organs to increase 3–4 times compared with the control mice. Administration of peptide FNIII14 completely inhibited both the metastatic foci and the increases in liver and spleen weight. RGD peptide also inhibited the increases in liver and spleen weight, but only weakly, whereas peptide FNIII14 exhibited inhibitory effects at lower molar concentration compared with RGD peptide [28]. Moreover, transplantation of mouse mammary tumors into the ventral flank of mice caused liver metastasis, and peptide FNIII14 co-administered with doxorubicin suppressed these metastases. Cheng and colleagues reported that dipeptidyl peptidase IV (DPPIV) binds to peptides containing the FN type III repeat region, and a peptide harboring FN type III repeat 14 (VTEATITGLEPGTEYTIY with the DPPIV-binding motif TITGLEPGTE) shows an anti-metastatic effect toward rat mammary carcinoma colonizing the lung by competitively blocking the adhesion between DPPIV and tumor cell surface-associated FN in a spontaneous metastasis mouse model [29]. Since integrin-inactivating peptide FNIII14 also contains the DPPIV-binding motif, further investigations are needed to uncover the undetermined mode of action of peptide FNIII14 for overcoming CAM-DR.
Frequent relapse after complete remission of acute myelogenous leukemia (AML) is an important clinical challenge in the treatment of leukemia. Relapse has been ascribed to minimal residual disease (MRD) in the bone marrow, and thus treatment strategies for the eradication of MRD in bone marrow are urgently required. The integrin-mediated adhesive interaction of leukemia cells with ECM confers resistance to anti-cancer drugs, referred to as cell adhesion-mediated drug resistance (CAM-DR). Peptide FNIII14 overcame CAM-DR to cytosine arabinoside (Ara C) in both AML cell lines and fresh patient-derived AML cells that adhered to FN by inactivating β1-integrin in vitro [30]. Treatment with a combination of FNIII14 and Ara C completely recovered the survival rate during the observation period in a mouse model of MRD in AML, and no AML cells were detected in organs, including bone marrow, suggesting that peptide FNIII14 successfully eradicated MRD in AML mice [30]. CAM-DR is associated with resistance to anti-cancer drugs for both hematological malignancies and solid cancers. Nakagawa and colleagues reported that adhesion to FN via β1-integrin conferred CAM-DR against 5-fluorouracil (5-FU) in oral squamous cell carcinoma (OSCC) cells, but peptide FNIII14 enhanced chemosensitivity to 5-FU by inhibiting cell adhesion in 5-FU-resistant OSCC cells, suggesting that peptide FNIII14 overcame CAM-DR [31]. Taken together, the application of FNIII14 in combination with chemotherapy could represent a promising therapeutic strategy.
To date, many competitive antagonist peptides for integrins derived from FN functional sequences have been widely investigated in preclinical and clinical studies as anti-cancer agents. Despite the demonstration of therapeutic potential in preclinical studies, numerous clinical studies have failed to show significant therapeutic benefits [32]. Peptide FNIII14 impedes integrin signaling by a mechanism entirely distinct from that of integrin competitive antagonists: unlike antagonistic peptides, peptide FNIII14 can induce conformational change in β1-integrin from the active to the inactive state. Our previous study showed that peptide FNIII14 can accelerate the death of glioblastoma cells in suspension culture (i.e., anchorage-independent conditions, through activation of the caspase signaling pathway). In contrast, peptides RGD and CS-1, which antagonize integrins α5β1, αvβ3, and α4β1, showed no such an effect [21]. Thus, peptide FNIII14 exhibits anti-cancer effects that cannot be achieved with conventional integrin antagonist peptides that competitively inhibit integrin-ECM binding.
Table 1. Anti-cancer activity of integrin-inactivating peptide FNIII14 in vitro and in vivo.
|
|
Cancer Type |
Cells/Experimental Model |
Ref. |
|
|
Suppression of survival/proliferation |
|
|||
|
In vitro |
Glioma/Glioblastoma |
T98G, 9L |
[20] |
|
|
|
Neuroblastoma |
IMR-32 |
[27] |
|
|
|
Pancreatic cancer |
MIA-PaCa 2 |
[27] |
|
|
|
Basophilic leukemia |
RBL-2H3 |
[33] |
|
|
In vivo |
Glioma/Glioblastoma |
Subcutaneous xenograft model |
[20] |
|
|
|
Neuroblastoma |
Subcutaneous xenograft model |
[27] |
|
|
Disruption of anoikis resistance |
|
|||
|
In vitro |
Glioma/Glioblastoma |
T98G, 9L |
[21] |
|
|
Suppression of migration/invasion |
|
|||
|
In vitro |
Glioma/Glioblastoma |
T98G |
[20] |
|
|
|
Lymphoma |
L5178Y-ML25 |
[28] |
|
|
|
Neuroblastoma |
IMR-32 |
[27] |
|
|
Suppression of Myc expression |
|
|||
|
In vitro |
Lung cancer |
NCI-H82 (c-myc) |
[27] |
|
|
|
Pancreatic cancer |
MIA-PaCa 2 (c-myc) |
[27] |
|
|
|
Chronic myelogenous leukemia |
K562 (c-myc) |
[27] |
|
|
|
Neuroblastoma |
IMR-32, NB-1, KELLY (N-myc) |
[27] |
|
|
Potentiation of anti-cancer activity |
|
|||
|
In vitro |
Glioma/Glioblastoma |
T98G, 9L, U251 (cotreatment with temozolomide) |
[8,20] |
|
|
|
Oral squamous cell carcinoma |
Ca9-22/FR2 (cotreatment with 5-fluorouracil) |
[31] |
|
|
|
Breast cancer |
4T1 (cotreatment with doxorubicin) |
[34] |
|
|
|
Melanoma |
B16BL6 (cotreatment with aclarubicin, vinblastine, 5-fluorouracil, and dacarbazine) |
[34] |
|
|
In vivo |
Glioma/Glioblastoma |
Subcutaneous xenograft model (cotreatment with temozolomide) |
[20] |
|
|
Inhibition of metastases |
|
|||
|
In vivo |
Breast cancer (liver metastases) |
Mouse model of experimental tumor metastasis |
[34] |
|
|
|
||||
|
|
Lymphoma (liver and spleen metastases) |
Mouse model of experimental tumor metastasis |
[28] |
|
|
Disruption of cell adhesion-mediated drug resistance to cytosine arabinoside |
|
|||
|
In vitro |
Acute myelogenous leukemia (AML) |
U937, HL-60, cells from AML patients |
[30] |
|
|
In vivo |
AML |
Mouse model of minimal residual disease in AML |
[30] |
|
|
Suppression of tumor onset |
|
|||
|
In vivo |
Colitis-associated colorectal cancer |
Azoxymethane-dextran sodium sulfate mouse model |
[35] |
|
T98G, human glioblastoma cell line; 9L, rat gliosarcoma cell line; IMR-32, human neuroblastoma cell line; MIA-PaCa 2, human pancreatic carcinoma cell line; RBL-2H3, rat basophilic leukemia cell line; L5178Y-ML25, murine T lymphoma cells; NCI-H82, human small cell lung cancer cell line; K562, human chronic myelogenous leukemia cell line; NB-1, human neuroblastoma cell line; KELLY, human neuroblastoma cell line; U251, human glioblastoma cell line; Ca9-22/FR2, 5-FU-resistant OSCC cell line; 4T1, mouse mammary tumor cell line; B16BL6, mouse melanoma cell line; U937, human monoblastic leukemia cell line; HL-60, human promyelocytic leukemia cell line. Table modified and revised from [8].
2.5. Perspectives
One drawback of peptide drugs is their low stability in vivo due to their susceptibility to proteolytic enzymes [36]. Peptide FNIII14 has been found to be easily degraded by aminopeptidases in the blood plasma. To impart resistance to serum aminopeptidases, the N-terminal E (Glu) amino acid residue of FNIII14 was replaced with the non-natural D-form (T-[D-Glu]-ATITGLEPGTEYTIYVIAL). D-form peptide FNIII14 is not recognized by these peptidases, in which leads to improved stability of the peptide in human blood. Furthermore, peptide FNIII14 has been confirmed to be non-myelotoxic in mice (1 mg intravenous route), and repeated administration of peptide FNIII14 did not affect body weight in mice [20,27,30]. The possible application of FNIII14 to human clinical practice requires further pharmacokinetic/pharmacodynamic analyses and non-clinical toxicity studies examining its genotoxicity and carcinogenicity.
Pathological accumulation of ECM proteins is a hallmark of fibrotic diseases and cancer [1,37]. Recent compelling evidence indicates that increased ECM stiffening and enhanced integrin-related aberrant mechano-signaling lead to the malignant progression of fibrosis and cancer [38–40]. Insightful studies have demonstrated the role of tenascin-C (TNC) in increased ECM stiffness in fibrotic areas or in tumor microenvironments [41–43]. For example, glioblastoma aggression correlates with increased TNC-enriched ECM stiffness and enhanced integrin-related aberrant mechano-signaling [41]. Moreover, a GBM xenograft model derived from cells expressing a conditional V737N β1-integrin auto-clustering active mutant exhibited enhanced mechano-signaling, increased TNC-enriched ECM stiffness, and subsequent enhanced tumor burden [42]. We previously found that the FN type III repeat A2 of TNC molecule has a cryptic site, called TNIIIA2, and TNC fragments/peptides containing this cryptic site can activate β1-integrins to strengthen cell adhesion to the ECM and sustain it for long periods of time [44,45]. Based on this unique activity, we speculate that this abnormal activation of β1-integrins may be a key event in the acquisition of aggressive properties, such as excessive survival/proliferation and disseminated migration [20,21,46]. Interestingly, TNIIIA2-induced aggressive properties can be inhibited by peptide FNIII14 through the inactivation of β1-integrins in glioblastoma cells [20]. TNC is highly expressed in several fibrotic organs and cancers, including glioblastoma, and especially mesenchymal glioblastoma, and its expression is therefore likely to be closely associated with a poor clinical outcome [47,48]. In addition, for several disorders other than those introduced in this entry, it is presumed that peptide FNIII14 may exert significant therapeutic effects.
3. Conclusions
FN-derived peptide FNIII14 is a new type of integrin-targeted agent that induces a conformational change in β1-integrin from the active to an inactive state, causing functional inactivation, and provides a promising therapeutic effect that cannot be achieved by conventional integrin competitive antagonists.