The ubiquitin proteasome system (UPS) governs the non-lysosomal degradation of oxidized, damaged, or misfolded proteins in eukaryotic cells. This process is tightly regulated through the activation and transfer of polyubiquitin chains to target proteins which are then recognized and degraded by the 26S proteasome complex. The role of UPS is crucial in regulating protein levels through degradation to maintain fundamental cellular processes such as growth, division, signal transduction, and stress response. Dysregulation of the UPS, resulting in loss of ability to maintain protein quality through proteolysis, is closely related to the development of various malignancies and tumorigenesis.
- ubiquitin proteasome system
- dysregulation
- chemoresistance
- cancer
- therapy
- inhibitors
1. The Ubiquitin Proteasome System
The ubiquitin proteasome system (UPS) is essential for the regulation of protein homeostasis and control of eukaryotic cellular processes including cell cycle progression, stress response, signal transduction, and transcriptional activation [1][2]. UPS controls the degradation of approximately 80% of intracellular proteins which are oxidized, damaged, or misfolded in eukaryotic cells [3]. Though the UPS and autophagy are both important systems of degradation of proteins, the sizes of substrates critically influence the choice of degradation pathway [4]. The UPS typically degrades single unfolded polypeptides, whereas autophagy deals with larger cytosolic complexes, cellular aggregates, and organelles.
Degradation of targeted proteins involves a tightly coordinated process where ubiquitin is covalently attached to the substrate protein through the sequential action of three enzymes. Ubiquitin is a small protein comprising 76 amino acids found in all eukaryotic cells [5]. The energy derived from ATP hydrolysis initiates the activation of ubiquitin activating enzyme (E1) allowing the formation of thioester bond between E1 and ubiquitin. This is followed by transfer of ubiquitin from E1 to ubiquitin-conjugating enzyme (E2), forming a thioester bond similar to that of E1. The third final step involves the covalent attachment of ubiquitin to lysine residues of target protein, catalyzed by ubiquitin ligase (E3) [6]. The 26S proteasome complex comprises a core 20S proteasome and one or two units of the regulatory 19S proteasome (Figure 1). Once a target protein has been modified with a polyubiquitin chain, it is recognized by the 19S proteosome which removes the polyubiquitin chain and the protein is then unfolded and translocated into the 20S proteasome where it is degraded into short peptides [7]. While polyubiquitination has been associated with protein clearance through proteasomal degradation, mono-ubiquitination, which involves the addition of a single ubiquitin moiety to the substrate protein, is shown to affect a range of cellular processes including kinase activity, epigenetic regulation, protein translocation, and DNA damage signaling [8][9].
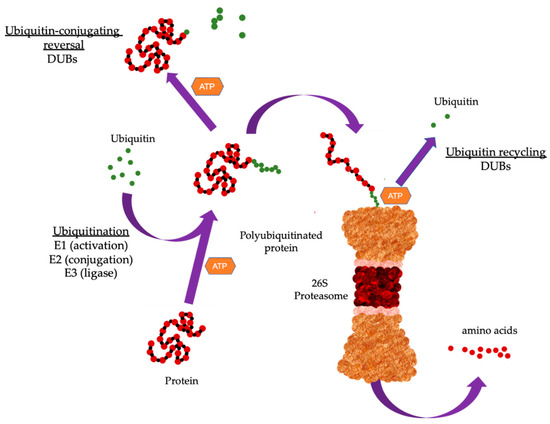
Figure 1. Overview of the ubiquitin proteasome system (UPS). The UPS cascade. Substrate protein is ubiquitinated through the sequential action of three enzymes. E1 binds to activated ubiquitin and is transferred to the ubiquitin-conjugating enzyme (E2). The E2 carries the activated ubiquitin to ubiquitin ligase (E3), which then facilitates the transfer of ubiquitin from E2 to a lysine residue in the target protein. Proteins can be modified with a single mono-ubiquitin molecule, or with ubiquitin chains of different lengths and linkage types. Substrate proteins modified with specific chains are recognized and subsequently degraded by the 26S proteasome. Deubiquitinating enzymes (DUBs) remove ubiquitin from substrate proteins by removing mono-ubiquitination or by trimming or removing the ubiquitin chain. Typically, poly-ubiquitination has been associated with protein clearance through proteasomal degradation while mono-ubiquitination which involves the addition of a single ubiquitin moiety to the substrate protein affects cellular processes.
Ubiquitin contains seven important lysine residues which can be ubiquitinated (K6, K11, K27, K33, K48, and K63) and can form polyubiquitin chains. The two best characterized ubiquitin linkages are K48 and K63 where K48 polyubiquitination targets proteins for degradation by the 26S proteasome complex [10] and K63 participates in DNA damage signaling and recruits DNA repair proteins to damage sites [11]. Protein ubiquitination can be reversed through the removal of ubiquitin from target proteins by deubiquitinating enzymes (DUBs), and this rescues protein destined for degradation. DUBs have also been implicated in the maturation, recycling, and editing of ubiquitin [12][13][14]. Further, chain configuration and linkage can endow ubiquitin with additional roles through the formation of more complex topologies with unknown activities [15]. Dysregulation or abnormal UPS function is frequently seen in various human malignancies and this identifies the aberrant components of the UPS as potential drug targets [16][17]. This review endeavors to present recent literature on the functional roles of UPS in human cancers. We cover how the dysregulation of UPS components may function either as an oncogene or tumor suppressor and affects cellular signaling in tumors. Further, we present current small inhibitors against the UPS and highlight issues that has severely restricted its development.
Increasing evidence demonstrate ubiquitin enzymes are important in carcinogenesis. Though there are numerous cancer-related studies on these enzymes, a large majority primarily focuses on E3 ligases. Studies on E1-activating enzymes have been largely used to identify potential targets in UPS inhibition in cancer while studies on E2-conjugated enzymes revealed their involvement in cell cycle progression, DNA repair, and regulation of oncogenic signaling pathways during tumorigenesis [18][19]. Further E2 enzymes are often found upregulated and highly correlated with poor prognosis in various malignancies including the pancreas, lung, breast, skin, and thyroid [20]. Currently, eight E1s, > 40 E2s, and > 600 E3s have been identified in the human proteome [21].
2. Dysregulation of UPS in cancer
2.1. E2 enzymes. The ubiquitin-conjugating E2 family comprises > 40 members, and modulates protein stability and ubiquitination through the conjugation of ubiquitin to target proteins [22]. E2-conjugating enzymes are also found dysregulated in cancers and reported to be potent mediators contributing towards multiple tumorigenic processes including migration/invasion, proliferation, drug resistance, radiation resistance, cell cycle, apoptosis, and stimulation of oncogenic pathways. Examples of dysregulated E2 enzymes in cancer are summarized in Table 1
Table 1. Summary of the functions of E2 and E3 enzymes in human cancers described in this review.
| Family | Name | Role | Cancer Type | Function | Test Model | Reference |
|---|---|---|---|---|---|---|
| E2 | UBE2C | Oncogene | Gastric | Chromosomal stability, Proliferation, Migration, Invasion | In vitro, In vivo | [23] |
| Oncogene | Colon | Cell cycle, Proloferation | In vitro | [24] | ||
| Oncogene | Colorectal | Proliferation, Invasion | In vitro | [25] | ||
| Oncogene | Thyroid | Proliferation | In vitro | [26] | ||
| Oncogene | Breast | Proliferation, Drug resistance, Radiation resistance | In vitro | [27] | ||
| Oncogene | Liver | Proliferation, Drug resistance, Migration, Invasion | In vitro | [28] | ||
| Oncogene | Non-small cell lung | Drug resistance | In vitro | [29] | ||
| UBE2Q1 | Oncogene | Colorectal | Proliferation | [30] | ||
| Oncogene | Liver | p53 signaling, Cell cycle | In vitro | [31] | ||
| Oncogene | Breast | p53 signaling | In vitro | [32] | ||
| UBE2S | Oncogene | Endometrial | SOX6/β-catenin signaling, Proliferation | In vitro | [33] | |
| Oncogene | Lung adenocarcinoma | Proliferation, p53 signaling, Apoptosis | In vitro | [34] | ||
| Oncogene | Liver | p53 signaling, Cell cycle | In vitro | [35] | ||
| E3 | FBW7 | Tumor suppressor | Burkitt’s lymphoma | c-Myc signaling | In vitro | [36][37] |
| Tumor suppressor | Chronic myelogenous leukemia | c-Myc signaling | In vitro, In vivo | [38] | ||
| Lipogenesis | Lung, Melanoma, Thyroid, Cervical | mTORC2/SREBP1 signaling | In vitro | [39] | ||
| Tumor suppressor | T cell leukemia | Notch signaling | In vitro, In vivo | [40] | ||
| Tumor suppressor | Colorectal | c-Myc signaling, Cell cycle | In vitro | [41] | ||
| Tumor suppressor | Esophageal squamous cell | c-Myc signaling | In vitro | [42] | ||
| Tumor suppressor | Colorectal, Cervical, Ovarian, Non-small cell lung | Apoptosis (via Mcl1) | In vitro | [43] | ||
| MDM2 | Oncogene | Neuroblastoma | p53 signaling | In vitro, In vivo | [44] | |
| Oncogene | Cervical | Cell cycle, Apoptosis | In vitro | [45] | ||
| Oncogene | Liver | Metastasis, Drug response | In vitro, In vivo | [46] | ||
| Cdc20 | Oncogene | Breast | Metastasis, Drug response | In vitro | [47] | |
| Cdh1 | Tumor suppressor | Breast | Src signaling | In vitro | [48] | |
| β-TRCP | Tumor suppressor | Breast, Prostate | MTSS1 signaling | In vitro | [49] | |
| Oncogene | Lung | FOXN2 | In vitro, In vivo | [50] | ||
| Tumor suppressor | Papillary thyroid | VEGFR2 signaling | In vitro, In vivo | [51] | ||
| E6AP | Oncogene | Prostate | Radiation response | In vitro | [52] | |
| Oncogene | Prostate | p27 signaling | In vitro, In vivo | [53] | ||
| Oncogene | Prostate | Metastasis | In vitro, In vivo | [54] |
E3 ubiquitin ligases are a large family of enzymes that promote ubiquitin transfer to proteins or polyubiquitin chains [55]. E3 ligases play an important role in the ubiquitin- mediated proteolytic cascade and are classified into four main classes, according to their domain structure and substrate recognition. The four E3 classes are the homologous to the E3 ubiquitin ligase E6-associated protein (E6AP) C-terminus (HECT), really interesting new gene (RING)-finger, U-box, and plant homeodomain (PHD)-finger. Depending on the substrate targets, E3 ligases can function either as a tumor suppressor or oncogene and can participate in various cellular processes including cell cycle, apoptosis, drug response, metastasis, radiation response, and oncogenic signaling. Examples of dysregulation of E3 ligases in cancer are summarised in Table 1.
Currently, about >100 DUB genes have been identified where the biological functions for the majority are still unknown [56][57]. USPs are, by far, the largest class of DUBs, com- prising ~60 human proteases with most containing several domains apart from the catalytic domain. Conserved sequences among these proteases are restricted to the catalytic domain which is designated by the catalytic motif containing Cys, His, and Asp (or Asn) residues. Conserved catalytic domains are thought to be important for substrate specificity, catalytic activity regulation, and mediating protein–protein interaction to each USP. Dysregulated DUBs in cancer are hence potential drug targets. The challenge in the development of drugs is the difficulty in designing a specific inhibitor for a single DUB. 3D crystallography structures of catalytic domains of USP2, USP7, USP8, and USP14 reveal a remarkable structural conservation of their active site shared among these enzymes, thus providing evidence that the development of inhibitors may prove to be challenging [58][59][60]. Further, the crystal structures show that the catalytic domains are in inactive conformation prior ubiquitin binding suggesting an alternative target for intervention. Apart from deubiquitination, DUBs have been shown to modulate cellular processes in human malignancies including DNA damage response, oncogenic signaling cascades, drug resistance, apoptosis, cell cycle, immunomodulation, and invasion/migration. Examples of dysregulated DUBs in cancer are summarised in Table 2.
Table 2. Summary of the functions of DUB enzymes described in this review.
| Name | Role | Cancer Type | Function | Test model | Reference |
| BAP1 | Tumor suppressor | Lung, Osteosarcoma, Colon | DNA double-strand repair | In vitro | [61][62][63] |
| Tumor suppressor | Renal | Ferroptosis signaling | In vitro | [64] | |
| USP7 | Oncogene | Lung | p53 signaling | In vitro, in vivo | [65] |
| Oncogene | Cervical | Self-renewal; Foxp3 signaling | In vitro | [66] | |
| Oncogene | Non-small cell lung | Immune Response; Foxp3 signaling | In vitro | [67] | |
| USP22 | Oncogene | Lung | Cell Cycle | In vitro | [68] |
| Oncogene | Lung adenocarcinoma | EGFR-TKI resistance | In vitro, in vivo | [69] | |
| Oncogene | Colon | CCNB1 signaling | In vitro, in vivo | [70] | |
| Oncogene | Glioblastoma | KDM1A signaling | In vitro, in vivo | [71] | |
| UCHL1 | Oncogene | Breast | Drug resistance; Invasion/migration | In vitro | [72] |
| Ataxin 3 | Oncogene | Breast, Osteosarcoma, Cervical, Colorectal | DNA | In vitro | [73] |
| Oncogene | Testicular | mTOR/Akt signaling | In vitro | [74] | |
| PSMD11 | Oncogene | Cervical. Osteosarcoma | DNA damage response | In vitro | [75] |
| Oncogene | Lung, Prostate, Colorectal, Breast, Cervix | Cell cycle | In vitro | [76] | |
| Oncogene | Liver | E2F1 signaling | In vitro, in vivo | [77] | |
| A20 | Tumor suppressor | Colorectal | Apoptosis signaling | In vitro | [78] |
| Tumor suppressor | Diffuse large B-cell lymphoma | NF-kβ signaling | In vitro | [79] | |
| Tumor suppressor | Sarcoma | NF-kβ signaling | In vitro | [80] |
3. UPS Inhibitors in Cancer therapy
The progress in targeting the UPS has been slow and this delay has been attributed to the following reasons. First, most components of the ubiquitin system do not possess a well-defined catalytic pocket to allow binding of small inhibitors. Second, the ubiquitination process relies on the dynamic rearrangement of multiple protein–protein interactions that traditionally have been challenging to disrupt with small molecule inhibitors. Third, components of the UPS are shown to possess both oncogenic and tumor suppressor properties due to the complexity of their regulatory cellular processes. Despite these challenges, components of the UPS have been considered as attractive targets for cancer treatment. In the following sections, we introduce some inhibitors against components of the UPS that have been tested in preclinical and clinical studies as summarized in Table 3.
Table 3. Summary of UPS inhibitors which are FDA-approved and/or tested in clinical trials described in this review.
| Inhibitor | Target | Cancer Type | Clinical Trial | Reference |
|---|---|---|---|---|
| Bortezomib | Proteasomal inhibitor | Multiple myeloma, Mantle cell lymphoma, Leukemia, Neuroblastoma, Head and Neck, Thyroid, Hepatocellular |
FDA approved | www.clinicaltrials.gov [81][82][83][84][85][86][87][88] |
| Carfilzomib | Proteasomal inhibitor | Multiple myeloma, Lymphoma, Relapsed and/or refractory multiple myeloma, Leukemia, Lung, Thyroid, Refractory renal cell carcinoma |
FDA approved | www.clinicaltrials.gov [52][89][90][91][92][88] |
| Ixazomib | Proteasomal inhibitor | Multiple myeloma, Relapsed and/or refractory multiple myeloma, Lymphoma, Leukemia, Breast, Glioblastoma, Renal cell carcinoma, Hodgkin and T cell lymphoma |
FDA approved | www.clinicaltrials.gov [93] |
| Delanzomib | Proteasomal inhibitor | Non-Hodgkin’s lymphoma | Phase I | www.clinicaltrials.gov |
| Marizomib | Proteasomal inhibitor | Multiple myeloma, Advanced solid tumors | Phase I/II | www.clinicaltrials.gov |
| Oprozomib | Proteasomal inhibitor | Multiple myeloma, Glioma, Pancreatic, Lung, Melanoma, Lymphoma, Glipblastoma | Phase I/II/III | www.clinicaltrials.gov |
| MLN4924 | NAE and UBA1(E1) | Advanced malignant solid tumors, Melanoma, Hepatocellular, B cell lymphoma, Hematologic malignancies, Acute myelocytic leukemia |
Phase I/II/III | www.clinicaltrials.gov |
| TAK981 | SAE (E1) | B cell lymphoma, colorectal, non-Hodgkin’s, Advcnced/metasiatic solid tumors | Phase I/II | www.clinicaltrials.gov |
| TAS4464 | NAE (E1) | Multiple myeloma, non-Hodgkin lymphoma | Phase I/II | www.clinicaltrials.gov |
| SAR-405838 | MDM2 (E2) | Solid tumors | Phase I | www.clinicaltrials.gov [87][94] |
| CGM-097 | MDM2 (E2) | Advanced p53 wildtype solid tumors | Phase I | www.clinicaltrials.gov [95][96] |
| DS-3032b | MDM2 (E2) | Acute myelocytic leukemia | Phase I/II | www.clinicaltrials.gov [97][98] |
| Debio1143 (AT-406) | cIAP1/2 (E3) | Acute myeloid leukemia | Phase I | www.clinicaltrials.gov [99] |
| LC-161 | IAP (E3) | Advanced solid tumors | Phase I | www.clinicaltrials.gov [100] |
| Birinapant | IAP (E3) | Solid tumors | Phase I/II | www.clinicaltrials.gov [101] |
| Pimozide | USP1 | Glioma, Non-small cell lung cancer | FDA approced for Tourette’s syndrome; Preclinical | [102][103] |
| Mitoxantrone | USP11 | Metastatic crastrate -resistant prostate, Acute myeloid leukemia, Advanced breast cancer, non-Hodgkin’s lymphoma, Primary liver |
FDA approved | [104][105][106][107][108][109][110][111][112] |
4. Conclusion
Frequent aberrant UPS activity seen in human malignancies indicate that the proteasome and components of the UPS are attractive therapeutic targets. Targeting the proteasome, in the clinic, has achieved success with FDA-approved proteasome inhibitors such as bortezomib, carfilzomib and ixazomib. Being the last step in the UPS, the use of proteasome inhibitors has shown undesirable side-effects arising from the action of up-stream UPS components. This shows that there is an untapped potential for the devel-opment of drugs against other components of the UPS. Thus, ubiquitin activating steps, E2, E3 and DUBs can be exploited for inhibition [113][114]. Unfortunately, most of these inhibitors show good efficacy in culture models but less so in animal models and clinical trials [115][116][117]. Traditionally, the ubiquitin activating steps and degradation possess the greatest potential due to presence of well-defined activity pockets but face issues of substrate specificity. The other UPS components however do not possess defined pockets for targeting with small inhibitors. Hence, delay in the development of successful UPS inhibitors can be attributed to the lack of knowledge of target protein structures and identifiable activity pockets for inhibitor binding. Advances in technology such as computer-aided design, mass spectrophotometry and high throughput screening may aid in the identification of suitable candidates. Further, the occurrence of oncogenic signaling together with aberrant UPS activity may affect the success of future UPS inhibitors. Nevertheless, a greater effort is required to elucidate the functions of aberrant UPS at both preclinical and clinical levels to better understand their roles in human malignancies to develop alternative paradigms for therapeutic intervention.
This entry is adapted from the peer-reviewed paper 10.3390/cancers13071513
References
- Ciechanover, A. Intracellular protein degradation: From a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Best Pract. Res. Clin. Haematol. 2017, 30, 341–355.
- Myung, J.; Kim, K.B.; Crews, C.M. The ubiquitin-proteasome pathway and proteasome inhibitors. Med. Res. Rev. 2001, 21, 245–273.
- Yang, H.; Chen, X.; Li, K.; Cheaito, H.; Yang, Q.; Wu, G.; Liu, J.; Dou, Q.P. Repurposing old drugs as new inhibitors of the ubiquitin-proteasome pathway for cancer treatment. Semin. Cancer Biol. 2021, 68, 105–122.
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224.
- Nandi, D.; Tahiliani, P.; Kumar, A.; Chandu, D. The ubiquitin-proteasome system. J. Biosci. 2006, 31, 137–155.
- Fang, S.; Weissman, A.M. Ubiquitin-proteasome system. Cell. Mol. Life Sci. 2004, 61, 1546–1561.
- Adams, J. The proteasome: Structure, function, and role in the cell. Cancer Treat. Rev. 2003, 29 (Suppl. S1), 3–9.
- Wang, G.; Gao, Y.; Li, L.; Jin, G.; Cai, Z.; Chao, J.-I.; Lin, H.-K. K63-Linked Ubiquitination in Kinase Activation and Cancer. Front. Oncol. 2012, 2, 5.
- Miranda, M.; Sorkin, A. Regulation of Receptors and Transporters by Ubiquitination: New Insights into Surprisingly Similar Mechanisms. Mol. Interv. 2007, 7, 157–167.
- Hochstrasser, M. Ubiquitin-Dependent protein degradation. Annu. Rev. Genet. 1996, 30, 405–439.
- Panier, S.; Durocher, D. Regulatory ubiquitylation in response to DNA double-strand breaks. DNA Repair 2009, 8, 436–443.
- Kim, J.H.; Park, K.C.; Chung, S.S.; Bang, O.; Chung, C.H. Deubiquitinating Enzymes as Cellular Regulators. J. Biochem. 2003, 134, 9–18.
- Nijman, S.M.; Luna-Vargas, M.P.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.; Sixma, T.K.; Bernards, R. A Genomic and Functional Inventory of Deubiquitinating Enzymes. Cell 2005, 123, 773–786.
- Clague, M.J.; Barsukov, I.; Coulson, J.M.; Liu, H.; Rigden, D.J.; Urbé, S. Deubiquitylases From Genes to Organism. Physiol. Rev. 2013, 93, 1289–1315.
- Komander, D. The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 2009, 37 Pt 5, 937–953.
- Ciechanover, A.; Brundin, P. The Ubiquitin Proteasome System in Neurodegenerative Diseases. Neuron 2003, 40, 427–446.
- Ciechanover, A.; Schwartz, A.L. The ubiquitin system: Pathogenesis of human diseases and drug targeting. Biochim. Biophys. Acta 2004, 1695, 3–17.
- Yang, Y.; Kitagaki, J.; Dai, R.-M.; Tsai, Y.C.; Lorick, K.L.; Ludwig, R.L.; Pierre, S.A.; Jensen, J.P.; Davydov, I.V.; Oberoi, P.; et al. Inhibitors of Ubiquitin-Activating Enzyme (E1), a New Class of Potential Cancer Therapeutics. Cancer Res. 2007, 67, 9472–9481.
- Liu, J.; Shaik, S.; Dai, X.; Wu, Q.; Zhou, X.; Wang, Z.; Wei, W. Targeting the ubiquitin pathway for cancer treatment. Biochim. Biophys. Acta 2015, 1855, 50–60.
- Hosseini, S.M.; Okoye, I.; Chaleshtari, M.G.; Hazhirkarzar, B.; Mohamadnejad, J.; Azizi, G.; Hojjat-Farsangi, M.; Mohammadi, H.; Shotorbani, S.S.; Jadidi-Niaragh, F. E2 ubiquitin-conjugating enzymes in cancer: Implications for immunotherapeutic interventions. Clin. Chim. Acta 2019, 498, 126–134.
- Clague, M.J.; Heride, C.; Urbé, S. The demographics of the ubiquitin system. Trends Cell Biol. 2015, 25, 417–426.
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 enzymes: More than just middle men. Cell Res. 2016, 26, 423–440.
- Zhang, J.; Liu, X.; Yu, G.; Liu, L.; Wang, J.; Chen, X.; Bian, Y.; Ji, Y.; Zhou, X.; Chen, Y.; et al. UBE2C Is a Potential Biomarker of Intestinal-Type Gastric Cancer With Chromosomal Instability. Front. Pharmacol. 2018, 9, 847.
- Fujita, T.; Ikeda, H.; Taira, N.; Hatoh, S.; Naito, M.; Doihara, H. Overexpression of UbcH10 alternates the cell cycle profile and accelerate the tumor proliferation in colon cancer. BMC Cancer 2009, 9, 87.
- Chen, S.; Chen, Y.; Hu, C.; Jing, H.; Cao, Y.; Liu, X. Association of clinicopathological features with UbcH10 expression in colorectal cancer. J. Cancer Res. Clin. Oncol. 2009, 136, 419–426.
- Pallante, P.; Berlingieri, M.; Troncone, G.; Kruhoffer, M.; Orntoft, T.; Viglietto, G.; Caleo, A.; Migliaccio, I.; Decaussin-Petrucci, M.; Santoro, M.; et al. UbcH10 overexpression may represent a marker of anaplastic thyroid carcinomas. Br. J. Cancer 2005, 93, 464–471.
- Rawat, A.; Gopal, G.; Selvaluxmy, G.; Rajkumar, T. Inhibition of ubiquitin conjugating enzyme UBE2C reduces proliferation and sensitizes breast cancer cells to radiation, doxorubicin, tamoxifen and letrozole. Cell. Oncol. 2013, 36, 459–467.
- Xiong, Y.; Lu, J.; Fang, Q.; Lu, Y.; Xie, C.; Wu, H.; Yin, Z. UBE2C functions as a potential oncogene by enhancing cell proliferation, migration, invasion, and drug resistance in hepatocellular carcinoma cells. Biosci. Rep. 2019, 39, BSR20182384.
- Guo, J.; Jin, D.; Wu, Y.; Yang, L.; Du, J.; Gong, K.; Chen, W.; Dai, J.; Miao, S.; Xi, S. The miR 495-UBE2C-ABCG2/ERCC1 axis reverses cisplatin resistance by downregulating drug resistance genes in cisplatin-resistant non-small cell lung cancer cells. EBioMedicine 2018, 35, 204–221.
- Fahmidehkar, M.A.; Shafiee, S.M.; Eftekhar, E.; Mahbudi, L.; Seghatoleslam, A. Induction of cell proliferation, clonogenicity and cell accumulation in S phase as a consequence of human UBE2Q1 overexpression. Oncol. Lett. 2016, 12, 2169–2174.
- Chang, R.; Wei, L.; Lu, Y.; Cui, X.; Lu, C.; Liu, L.; Jiang, D.; Xiong, Y.; Wang, G.; Wan, C.; et al. Upregulated expression of ubiquitin-conjugating enzyme E2Q1 (UBE2Q1) is associated with enhanced cell proliferation and poor prognosis in human hapatocellular carcinoma. J. Mol. Histol. 2014, 46, 45–56.
- Shafiee, S.M.; Rasti, M.; Seghatoleslam, A.; Azimi, T.; Owji, A.A. UBE2Q1 in a Human Breast Carcinoma Cell Line: Overexpression and Interaction with p53. Asian Pac. J. Cancer Prev. 2015, 16, 3723–3727.
- Lin, M.; Lei, T.; Zheng, J.; Chen, S.; Du, L.; Xie, H. UBE2S mediates tumor progression via SOX6/β-Catenin signaling in endometrial cancer. Int. J. Biochem. Cell Biol. 2019, 109, 17–22.
- Liu, Z.; Xu, L. UBE2S promotes the proliferation and survival of human lung adenocarcinoma cells. BMB Rep. 2018, 51, 642–647.
- Pan, Y.-H.; Yang, M.; Liu, L.-P.; Wu, D.-C.; Li, M.-Y.; Su, S.-G. UBE2S enhances the ubiquitination of p53 and exerts oncogenic activities in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2018, 503, 895–902.
- Bahram, F.; von der Lehr, N.; Cetinkaya, C.; Larsson, L.G. c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood 2000, 95, 2104–2110.
- Yeh, C.-H.; Bellon, M.; Nicot, C. FBXW7: A critical tumor suppressor of human cancers. Mol. Cancer 2018, 17, 115.
- Reavie, L.; Buckley, S.M.; Loizou, E.; Takeishi, S.; Aranda-Orgilles, B.; Ndiaye-Lobry, D.; Abdel-Wahab, O.; Ibrahim, S.; Nakayama, K.I.; Aifantis, I. Regulation of c-Myc Ubiquitination Controls Chronic Myelogenous Leukemia Initiation and Progression. Cancer Cell 2013, 23, 362–375.
- Li, S.; Oh, Y.T.; Yue, P.; Khuri, F.R.; Sun, S.-Y. Inhibition of mTOR complex 2 induces GSK3/FBXW7-dependent degradation of sterol regulatory element-binding protein 1 (SREBP1) and suppresses lipogenesis in cancer cells. Oncogene 2016, 35, 642–650.
- Yeh, C.-H.; Bellon, M.; Pancewicz-Wojtkiewicz, J.; Nicot, C. Oncogenic mutations in the FBXW7 gene of adult T-cell leukemia patients. Proc. Natl. Acad. Sci. USA 2016, 113, 6731–6736.
- Iwatsuki, M.; Mimori, K.; Ishii, H.; Yokobori, T.; Takatsuno, Y.; Sato, T.; Toh, H.; Onoyama, I.; Nakayama, K.I.; Baba, H.; et al. Loss of FBXW7, a cell cycle regulating gene, in colorectal cancer: Clinical significance. Int. J. Cancer 2010, 126, 1828–1837.
- Mori, M.; Yokobori, T.; Mimori, K.; Iwatsuki, M.; Ishii, H.; Tanaka, F.; Sato, T.; Toh, H.; Sudo, T.; Iwaya, T.; et al. Copy number loss of FBXW7 is related to gene expression and poor prognosis in esophageal squamous cell carcinoma. Int. J. Oncol. 2012, 41, 253–259.
- Wertz, I.E.; Kusam, S.; Lam, C.; Okamoto, T.; Sandoval, W.; Anderson, D.J.; Helgason, E.; Ernst, J.A.; Eby, M.; Liu, J.; et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011, 471, 110–114.
- Wang, W.; Wang, X.; Rajaei, M.; Youn, J.Y.; Zafar, A.; Deokar, H.; Buolamwini, J.K.; Yang, J.; Foster, J.H.; Zhou, J.; et al. Targeting MDM2 for Neuroblastoma Therapy: In Vitro and In Vivo Anticancer Activity and Mechanism of Action. Cancers 2020, 12, 3651.
- Xu, L.; Wang, J.; Yuan, X.; Yang, S.; Xu, X.; Li, K.; He, Y.; Wei, L.; Zhang, J.; Tian, Y. IU1 suppresses proliferation of cervical cancer cells through MDM2 degradation. Int. J. Biol. Sci. 2020, 16, 2951–2963.
- Wang, W.; Hu, B.; Qin, J.-J.; Cheng, J.-W.; Li, X.; Rajaei, M.; Fan, J.; Yang, X.-R.; Zhang, R. A novel inhibitor of MDM2 oncogene blocks metastasis of hepatocellular carcinoma and overcomes chemoresistance. Genes Dis. 2019, 6, 419–430.
- Cheng, S.; Castillo, V.; Sliva, D. CDC20 associated with cancer metastasis and novel mushroom-derived CDC20 inhibitors with antimetastatic activity. Int. J. Oncol. 2019, 54, 2250–2256.
- Han, T.; Jiang, S.; Zheng, H.; Yin, Q.; Xie, M.; Little, M.R.; Yin, X.; Chen, M.; Song, S.J.; Beg, A.A.; et al. Interplay between c-Src and the APC/C co-activator Cdh1 regulates mammary tumorigenesis. Nat. Commun. 2019, 10, 3716.
- Zhong, J.; Shaik, S.; Wan, L.; Tron, A.E.; Wang, Z.; Sun, L.; Inuzuka, H.; Wei, W. SCFβ-TRCP targets MTSS1 for ubiquitination-mediated destruction to regulate cancer cell proliferation and migration. Oncotarget 2013, 4, 2339–2353.
- Ma, J.; Lu, Y.; Zhang, S.; Li, Y.; Huang, J.; Yin, Z.; Ren, J.; Huang, K.; Liu, L.; Yang, K.; et al. β-Trcp ubiquitin ligase and RSK2 kinase-mediated degradation of FOXN2 promotes tumorigenesis and radioresistance in lung cancer. Cell Death Differ. 2018, 25, 1473–1485.
- Shaik, S.; Nucera, C.; Inuzuka, H.; Gao, D.; Garnaas, M.; Frechette, G.; Harris, L.; Wan, L.; Fukushima, H.; Husain, A.; et al. SCFβ-TRCP suppresses angiogenesis and thyroid cancer cell migration by promoting ubiquitination and destruction of VEGF receptor 2. J. Exp. Med. 2012, 209, 1289–1307.
- Paul, P.J.; Raghu, D.; Chan, A.-L.; Gulati, T.; Lambeth, L.; Takano, E.; Herold, M.J.; Hagekyriakou, J.; Vessella, R.L.; Fedele, C.; et al. Restoration of tumor suppression in prostate cancer by targeting the E3 ligase E6AP. Oncogene 2016, 35, 6235–6245.
- Raghu, D.; Paul, P.J.; Gulati, T.; Deb, S.; Khoo, C.; Russo, A.; Gallo, E.; Blandino, G.; Chan, A.-L.; Takano, E.; et al. E6AP promotes prostate cancer by reducing p27 expression. Oncotarget 2017, 8, 42939–42948.
- Gamell, C.; Bandilovska, I.; Gulati, T.; Kogan, A.; Lim, S.C.; Kovacevic, Z.; Takano, E.A.; Timpone, C.; Agupitan, A.D.; Litchfield, C.; et al. E6AP Promotes a Metastatic Phenotype in Prostate Cancer. iScience 2019, 22, 1–15.
- Michael J. Clague; Claire Heride; Sylvie Urbé; The demographics of the ubiquitin system. Trends in Cell Biology 2015, 25, 417-426, 10.1016/j.tcb.2015.03.002.
- Sebastian M.B. Nijman; Mark P.A. Luna-Vargas; Arno Velds; Thijn R. Brummelkamp; Annette M.G. Dirac; Titia K. Sixma; René Bernards; A Genomic and Functional Inventory of Deubiquitinating Enzymes. Cell 2005, 123, 773-786, 10.1016/j.cell.2005.11.007.
- Michael J. Clague; Igor Barsukov; Judy M. Coulson; Han Liu; Daniel J. Rigden; Sylvie Urbé; Deubiquitylases From Genes to Organism. Physiological Reviews 2013, 93, 1289-1315, 10.1152/physrev.00002.2013.
- Min Hu; Pingwei Li; Muyang Li; Wenyu Li; Tingting Yao; Jia-Wei Wu; Wei Gu; Robert E. Cohen; Yigong Shi; Crystal Structure of a UBP-Family Deubiquitinating Enzyme in Isolation and in Complex with Ubiquitin Aldehyde. Cell 2002, 111, 1041-1054, 10.1016/s0092-8674(02)01199-6.
- Min Hu; Pingwei Li; Ling Song; Philip D Jeffrey; Tatiana A Chernova; Keith D Wilkinson; Robert E Cohen; Yigong Shi; Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. The EMBO Journal 2005, 24, 3747-3756, 10.1038/sj.emboj.7600832.
- Martin Renatus; Shirley Gil Parrado; Allan D'Arcy; Ulf Eidhoff; Bernd Gerhartz; Ulrich Hassiepen; Benoit Pierrat; Ralph Riedl; Daniela Vinzenz; Susanne Worpenberg; et al. Structural Basis of Ubiquitin Recognition by the Deubiquitinating Protease USP2. Structure 2006, 14, 1293-1302, 10.1016/j.str.2006.06.012.
- Ismail Hassan Ismail; Riley Davidson; Jean-Philippe Gagné; Zhi Zhong Xu; Guy G. Poirier; Michael J. Hendzel; Germline Mutations in BAP1 Impair Its Function in DNA Double-Strand Break Repair. Cancer Research 2014, 74, 4282-4294, 10.1158/0008-5472.can-13-3109.
- Helen Yu; Helen Pak; Ian Hammond-Martel; Mehdi Ghram; Amélie Rodrigue; Salima Daou; Haithem Barbour; Luc Corbeil; Josée Hébert; Elliot Drobetsky; et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proceedings of the National Academy of Sciences 2013, 111, 285-290, 10.1073/pnas.1309085110.
- Han-Sae Lee; Hye-Ran Seo; Shin-Ai Lee; Soohee Choi; Dongmin Kang; Jongbum Kwon; BAP1 promotes stalled fork restart and cell survival via INO80 in response to replication stress. Biochemical Journal 2019, 476, 3053-3066, 10.1042/bcj20190622.
- Yilei Zhang; Jiejun Shi; Xiaoguang Liu; Li Feng; Zihua Gong; Pranavi Koppula; Kapil Sirohi; Xu Li; Yongkun Wei; Hyemin Lee; et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nature 2018, 20, 1181-1192, 10.1038/s41556-018-0178-0.
- Muyang Li; Delin Chen; Ariel L Shiloh; Jianyuan Luo; Anatoly Y. Nikolaev; Jun Qin; Wei Gu; Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 2002, 416, 648-653, 10.1038/nature737.
- Jorg van Loosdregt; Paul J. Coffer; Post-translational modification networks regulating FOXP3 function. Trends in Immunology 2014, 35, 368-378, 10.1016/j.it.2014.06.005.
- Lili Wang; Ruilin Guan; Lina Xie; Xinxing Liao; Kai Xiong; Thomas W. Rees; Yu Chen; Liangnian Ji; Hui Chao; An ER‐Targeting Iridium(III) Complex That Induces Immunogenic Cell Death in Non‐Small‐Cell Lung Cancer. Angewandte Chemie International Edition 2020, 60, 4657-4665, 10.1002/anie.202013987.
- Xiao-Yong Zhang; Maya Varthi; Stephen M. Sykes; Charles Phillips; Claude Warzecha; Wenting Zhu; Anastasia Wyce; Alan W. Thorne; Shelley L. Berger; Steven B. McMahon; et al. The Putative Cancer Stem Cell Marker USP22 Is a Subunit of the Human SAGA Complex Required for Activated Transcription and Cell-Cycle Progression. Molecular Cell 2008, 29, 102-111, 10.1016/j.molcel.2007.12.015.
- Huijuan Zhang; Bing Han; Hailing Lu; Yanbin Zhao; Xuesong Chen; Qingwei Meng; Mengru Cao; Li Cai; Jing Hu; USP22 promotes resistance to EGFR-TKIs by preventing ubiquitination-mediated EGFR degradation in EGFR-mutant lung adenocarcinoma. Cancer Letters 2018, 433, 186-198, 10.1016/j.canlet.2018.07.002.
- Zhenghong Lin; Can Tan; Quan Qiu; Sinyi Kong; Heeyoung Yang; Fang Zhao; Zhaojian Liu; Jinping Li; Qingfei Kong; Beixue Gao; et al. Ubiquitin-specific protease 22 is a deubiquitinase of CCNB1. Cell Discovery 2015, 1, 15028, 10.1038/celldisc.2015.28.
- Aidong Zhou; Kangyu Lin; Sicong Zhang; Yaohui Chen; Nu Zhang; Jianfei Xue; Zhongyong Wang; Kenneth D. Aldape; Keping Xie; James R. Woodgett; et al. Nuclear GSK3β promotes tumorigenesis by phosphorylating KDM1A and inducing its deubiquitylation by USP22. Nature 2016, 18, 954-966, 10.1038/ncb3396.
- Wenjuan Wang; Liping Zou; Danmei Zhou; Zhongwen Zhou; Feng Tang; Zude Xu; Xiuping Liu; Overexpression of ubiquitin carboxyl terminal hydrolase-L1 enhances multidrug resistance and invasion/metastasis in breast cancer by activating the MAPK/Erk signaling pathway. Molecular Carcinogenesis 2015, 55, 1329-1342, 10.1002/mc.22376.
- Abhay Narayan Singh; Judith Oehler; Ignacio Torrecilla; Susan Kilgas; Shudong Li; Bruno Vaz; Claire Guérillon; John Fielden; Esperanza Hernandez‐Carralero; Elisa Cabrera; et al. The p97–Ataxin 3 complex regulates homeostasis of the DNA damage response E3 ubiquitin ligase RNF 8. The EMBO Journal 2019, 38, e102361, 10.15252/embj.2019102361.
- Zhan Shi; Jiaxin Chen; Xiangmin Zhang; Jian Chu; Zhitao Han; Da Xu; Sishun Gan; Xiuwu Pan; Jianqing Ye; Xingang Cui; et al. Ataxin-3 promotes testicular cancer cell proliferation by inhibiting anti-oncogene PTEN. Biochemical and Biophysical Research Communications 2018, 503, 391-396, 10.1016/j.bbrc.2018.06.047.
- Laura R. Butler; Ruth M. Densham; Junying Jia; Alexander J. Garvin; Helen R. Stone; Vandna Shah; Daniel Weekes; Frederic Festy; James Beesley; Joanna R. Morris; et al. The proteasomal de-ubiquitinating enzyme POH1 promotes the double-strand DNA break response. The EMBO Journal 2012, 31, 3918-3934, 10.1038/emboj.2012.232.
- Ann Byrne; Rajashree P. McLaren; Paul Mason; Lilly Chai; Michael R. Dufault; Yinyin Huang; Beirong Liang; Joseph D. Gans; Mindy Zhang; Kara Carter; et al. Knockdown of human deubiquitinase PSMD14 induces cell cycle arrest and senescence. Experimental Cell Research 2010, 316, 258-271, 10.1016/j.yexcr.2009.08.018.
- Boshi Wang; Aihui Ma; Li Zhang; Wei-Lin Jin; Xia Qiang; Guiqin Xu; Bijun Qiu; Zhaojuan Yang; Yun Liu; Qiang Xia; et al. POH1 deubiquitylates and stabilizes E2F1 to promote tumour formation. Nature Communications 2015, 6, 8704-8704, 10.1038/ncomms9704.
- Z Jin; Cullin3-Based Polyubiquitination and p62-Dependent Aggregation of Caspase-8 Mediate Extrinsic Apoptosis Signaling. Cell 2009, 137, 736-748, 10.1016/s9999-9994(09)00537-6.
- Sang-Woo Kim; Kumaraguruparan Ramasamy; Hakim Bouamar; An-Ping Lin; Daifeng Jiang; Ricardo C. T. Aguiar; MicroRNAs miR-125a and miR-125b constitutively activate the NF- B pathway by targeting the tumor necrosis factor alpha-induced protein 3 (TNFAIP3, A20). Proceedings of the National Academy of Sciences 2012, 109, 7865-7870, 10.1073/pnas.1200081109.
- Mumtaz Yaseen Balkhi; O. Hans Iwenofu; Nadine Bakkar; Katherine J. Ladner; Dawn S. Chandler; Peter J. Houghton; Cheryl A. London; William Kraybill; Danilo Perrotti; Carlo M. Croce; et al. miR-29 Acts as a Decoy in Sarcomas to Protect the Tumor Suppressor A20 mRNA from Degradation by HuR. Science Signaling 2013, 6, ra63-ra63, 10.1126/scisignal.2004177.
- Lu, Y.; Orr, A.; Everett, R.D. Stimulation of the Replication of ICP0-Null Mutant Herpes Simplex Virus 1 and pp71-Deficient Human Cytomegalovirus by Epstein-Barr Virus Tegument Protein BNRF1. J. Virol. 2016, 90, 9664–9673.
- Hideshima, T.; Richardson, P.; Chauhan, D.; Palombella, V.J.; Elliott, P.J.; Adams, J.; Anderson, K.C. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001, 61, 3071–3076.
- Chauhan, D.; Hideshima, T.; Mitsiades, C.; Richardson, P.; Anderson, K.C. Proteasome inhibitor therapy in multiple myeloma. Mol. Cancer Ther. 2005, 4, 686–692.
- Liu, F.-T.; Agrawal, S.G.; Gribben, J.G.; Ye, H.; Du, M.-Q.; Newland, A.C.; Jia, L. Bortezomib blocks Bax degradation in malignant B cells during treatment with TRAIL. Blood 2008, 111, 2797–2805.
- Pei, X.-Y.; Dai, Y.; Grant, S. Synergistic Induction of Oxidative Injury and Apoptosis in Human Multiple Myeloma Cells by the Proteasome Inhibitor Bortezomib and Histone Deacetylase Inhibitors. Clin. Cancer Res. 2004, 10, 3839–3852.
- Mata-Cantero, L.; Lobato-Gil, S.; Aillet, F.; Lang, V.; Rodriguez, M.S. The Ubiquitin-Proteasome System (UPS) as a Cancer Drug Target: Emerging Mechanisms and Therapeutics. In Stress Response Pathways in Cancer; Wondrak, G., Ed.; Springer: Dordrecht, The Netherlands, 2015; pp. 225–264.
- Zhang, X.; Linder, S.; Bazzaro, M. Drug Development Targeting the Ubiquitin–Proteasome System (UPS) for the Treatment of Human Cancers. Cancers 2020, 12, 902.
- Stewart, A.K.; Rajkumar, S.V.; Dimopoulos, M.A.; Masszi, T.; Špička, I.; Oriol, A.; Hájek, R.; Rosiñol, L.; Siegel, D.S.; Mihaylov, G.G.; et al. Carfilzomib, Lenalidomide, and Dexamethasone for Relapsed Multiple Myeloma. N. Engl. J. Med. 2015, 372, 142–152.
- Kortuem, K.M.; Stewart, A.K. Carfilzomib. Blood 2013, 121, 893–897.
- Meng, L.; Mohan, R.; Kwok, B.H.B.; Elofsson, M.; Sin, N.; Crews, C.M. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc. Nat. Acad. Sci. USA 1999, 96, 10403–10408.
- Demo, S.D.; Kirk, C.J.; Aujay, M.A.; Buchholz, T.J.; Dajee, M.; Ho, M.N.; Jiang, J.; Laidig, G.J.; Lewis, E.R.; Parlati, F.; et al. Antitumor Activity of PR-171, a Novel Irreversible Inhibitor of the Proteasome. Cancer Res. 2007, 67, 6383–6391.
- Arastu-Kapur, S.; Anderl, J.L.; Kraus, M.; Parlati, F.; Shenk, K.D.; Lee, S.J.; Muchamuel, T.; Bennett, M.K.; Driessen, C.; Ball, A.J.; et al. Nonproteasomal Targets of the Proteasome Inhibitors Bortezomib and Carfilzomib: A Link to Clinical Adverse Events. Clin. Cancer Res. 2011, 17, 2734–2743.
- Chauhan, D.; Tian, Z.; Zhou, B.; Kuhn, D.; Orlowski, R.; Raje, N.; Richardson, P.; Anderson, K.C. In Vitro and In Vivo Selective Antitumor Activity of a Novel Orally Bioavailable Proteasome Inhibitor MLN9708 against Multiple Myeloma Cells. Clin. Cancer Res. 2011, 17, 5311–5321.
- Pulvino, M.; Liang, Y.; Oleksyn, D.; DeRan, M.; Van Pelt, E.; Shapiro, J.; Sanz, I.; Chen, L.; Zhao, J. Inhibition of proliferation and sur- vival of diffuse large B-cell lymphoma cells by a small-molecule inhib-itor of the ubiquitin-conjugating enzyme Ubc13-Uev1A. Blood 2012, 120, 1668–1677.
- Bauer, S.; Demetri, G.; Jeay, S.; Dummer, R.; Guerreiro, N.; Tan, D.; Kumar, A.; Meille, C.; Van Bree, L.; Halilovic, E.; et al. A phase I, open-label, multi-center, dose escalation study of oral NVP-CGM097, a p53/HDM2-protein-protein interaction inhibitor, in adult patients with selected advanced solid tumors. Ann. Oncol. 2016, 27 (Suppl. S6), vi114–vi135.
- Holzer, P.; Masuya, K.; Furet, P.; Kallen, J.; Valat-Stachyra, T.; Ferretti, S.; Berghausen, J.; Bouisset-Leonard, M.; Buschmann, N.; Pissot-Soldermann, C.; et al. Discovery of a Dihydroisoquinolinone Derivative (NVP-CGM097): A Highly Potent and Selective MDM2 Inhibitor Undergoing Phase 1 Clinical Trials in p53wt Tumors. J. Med. Chem. 2015, 58, 6348–6358.
- Bauer, T.; Hong, D.; Somaiah, N.; Cai, C.; Song, S.; Kumar, P.; Gajee, R.; Rosen, M.; Kochan, J.; Chen, S.; et al. Abstract B27: A phase I dose escalation study of the MDM2 inhibitor DS-3032b in patients with advanced solid tumors and lymphomas. Mol. Cancer Ther. 2015, 14 (Suppl. S2), Abstract nr B27.
- Gounder, M.M.; Bauer, T.M.; Schwartz, G.K.; Masters, T.; Carvajal, R.D.; Song, S.; Kumar, P.; Gajee, R.; Zernovak, O.; Rosen, M.M.; et al. A phase 1 study of the MDM2 inhibitor DS-3032b in patients (pts) with advanced solid tumors and lymphomas. J. Clin. Oncol. 2016, 34 (Suppl. S15), 2581.
- DiPersio, J.F.; Erba, H.P.; Larson, R.A.; Luger, S.M.; Tallman, M.S.; Brill, J.M.; Vuagniaux, G.; Rouits, E.; Sorensen, J.M.; Zanna, C. Oral Debio1143 (AT406), an Antagonist of Inhibitor of Apoptosis Proteins, Combined With Daunorubicin and Cytarabine in Patients With Poor-Risk Acute Myeloid Leukemia—Results of a Phase I Dose-Escalation Study. Clin. Lymphoma Myeloma Leuk. 2015, 15, 443–449.
- Infante, J.R.; Dees, E.C.; Olszanski, A.J.; Dhuria, S.V.; Sen, S.; Cameron, S.; Cohen, R.B. Phase I Dose-Escalation Study of LCL161, an Oral Inhibitor of Apoptosis Proteins Inhibitor, in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2014, 32, 3103–3110.
- Schilder, R.J.; Albertella, M.; Strauss, J.; Sydvander, M.; Nair, S.M.; Urakpo, K.; Norin, S.; Öhd, J. A phase 1/2 study with birinapant in combination with pembrolizumab. J. Clin. Oncol. 2020, 36 (Suppl. S15), TPS3131.
- Chen, J.; Dexheimer, T.S.; Ai, Y.; Liang, Q.; Villamil, M.A.; Inglese, J.; Maloney, D.J.; Jadhav, A.; Simeonov, A.; Zhuang, Z. Selective and Cell-Active Inhibitors of the USP1/ UAF1 Deubiquitinase Complex Reverse Cisplatin Resistance in Non-small Cell Lung Cancer Cells. Chem. Biol. 2011, 18, 1390–1400.
- Rath, B.H.; Camphausen, K.; Tofilon, P.J. Glioblastoma radiosensitization by pimozide. Transl. Cancer Res. 2016, 5 (Suppl. S6), S1029–S1032.
- Green, A.K.; Corty, R.W.; Wood, W.A.; Meeneghan, M.; Reeder-Hayes, K.E.; Basch, E.; Milowsky, M.I.; Dusetzina, S.B. Comparative Effectiveness of Mitoxantrone Plus Prednisone Versus Prednisone Alone in Metastatic Castrate-Resistant Prostate Cancer After Docetaxel Failure. Oncolologist 2015, 20, 516–522.
- Abbi, K.K.; Rybka, W.; Ehmann, W.C.; Claxton, D.F. Phase I/II Study of Clofarabine, Etoposide, and Mitoxantrone in Patients with Refractory or Relapsed Acute Leukemia. Clin. Lymphoma Myeloma Leuk. 2015, 15, 41–46.
- Gill, H.; Yim, R.; Pang, H.H.; Lee, P.; Chan, T.S.Y.; Hwang, Y.; Leung, G.M.K.; Ip, H.; Leung, R.Y.Y.; Yip, S.; et al. Clofarabine, cytarabine, and mitoxantrone in refractory/relapsed acute myeloid leukemia: High response rates and effective bridge to allogeneic hematopoietic stem cell transplantation. Cancer Med. 2020, 9, 3371–3382.
- Cornbleet, M.A.; Stuart-Harris, R.C.; Smith, I.E.; Coleman, R.E.; Rubens, R.D.; McDonald, M.; Mouridsen, H.T.; Rainer, H.; Van Oosterom, A.T.; Smyth, J.F. Mitoxantrone for the treatment of advanced breast cancer: Single-agent therapy in previously untreated patients. Eur. J. Cancer Clin. Oncol. 1984, 20, 1141–1146.
- Neidhart, J.A.; Gochnour, D.; Roach, R.; Hoth, D.; Young, D. A comparison of mitoxantrone and doxorubicin in breast cancer. J. Clin. Oncol. 1986, 4, 672–677.
- Cristofanilli, M.; Holmes, F.A.; Esparza, L.; Valero, V.; Buzdar, A.U.; Neidhart, J.A.; Hortobagyi, G.N. Phase I/II trial of high dose mitoxantrone in metastatic breast cancer: The M.D. Anderson Cancer Center experience. Breast Cancer Res. Treat. 1999, 54, 225–233.
- Silver, R.T.; Case, D.C.; Wheeler, R.H.; Miller, T.P.; Stein, R.S.; Stuart, J.J.; Peterson, B.A.; Rivkin, S.E.; Golomb, H.M.; Costanzi. Multicenter clinical trial of mitoxantrone in non-Hodgkin’s lymphoma and Hodgkin’s disease. J. Clin. Oncol. 1991, 9, 754–761.
- Bajetta, E.; Buzzoni, R.; Valagussa, P.; Bonadonna, G. Mitoxantrone: An active agent in refractory non-Hodgkin’s lymphomas. Am. J. Clin. Oncol. 1988, 11, 100–103.
- Davis, R.B.; Van Echo, D.A.; Leone, L.A.; Henderson, E.S. Phase II trial of mitoxantrone in advanced primary liver cancer: A Cancer and Leukemia Group B Study. Cancer Treat. Rep. 1986, 70, 1125–1126.
- Aliaksandr Khaminets; Christian Behl; Ivan Dikic; Ubiquitin-Dependent And Independent Signals In Selective Autophagy. Trends in Cell Biology 2016, 26, 6-16, 10.1016/j.tcb.2015.08.010.
- Kristal Duncan; Georgia Schäfer; Akhona Vava; M Iqbal Parker; Luiz F Zerbini; Targeting neddylation in cancer therapy. Future Oncology 2012, 8, 1461-1470, 10.2217/fon.12.131.
- Fatemeh Fouladkou; Tamara Landry; Hiroshi Kawabe; Antje Neeb; Chen Lu; Nils Brose; Vuk Stambolic; Daniela Rotin; The ubiquitin ligase Nedd4-1 is dispensable for the regulation of PTEN stability and localization. Proceedings of the National Academy of Sciences 2008, 105, 8585-8590, 10.1073/pnas.0803233105.
- Xinjiang Wang; Lloyd C. Trotman; Theresa Koppie; Andrea Alimonti; Zhenbang Chen; Zhonghua Gao; Junru Wang; Hediye Erdjument-Bromage; Paul Tempst; Carlos Cordon-Cardo; et al. NEDD4-1 Is a Proto-Oncogenic Ubiquitin Ligase for PTEN. Cell 2007, 128, 129-139, 10.1016/j.cell.2006.11.039.
- Lloyd C. Trotman; Xinjiang Wang; Andrea Alimonti; Zhenbang Chen; Julie Teruya-Feldstein; Haijuan Yang; Nikola P. Pavletich; Brett S. Carver; Carlos Cordon-Cardo; Hediye Erdjument-Bromage; et al. Ubiquitination Regulates PTEN Nuclear Import and Tumor Suppression. Cell 2007, 128, 141-156, 10.1016/j.cell.2006.11.040.