Aging is inevitable and it is one of the major contributors to cognitive decline. However, the mechanisms underlying age-related cognitive decline are still the object of extensive research. At the biological level, it is unknown how the aging brain is subjected to progressive oxidative stress and neuroinflammation which determine, among others, mitochondrial dysfunction. The link between mitochondrial dysfunction and cognitive impairment is becoming ever more clear by the presence of significant neurological disturbances in human mitochondrial diseases. Possibly, the most important lifestyle factor determining mitochondrial functioning is nutrition.
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
Cognition, broadly defined as encompassing the mental processes of learning, reasoning and memory, depends on the brain’s ability to undergo functional (e.g., adjusting efficacy of synaptic transmission) and structural (e.g., forming new synapses) alterations in response to changes in its environment—a phenomenon known as neuroplasticity. Internal and external stimuli—such as new experiences and learning, trigger changes in neuronal activity which induce re-organisation of neuronal networks and fine-tuning of brain circuitry, with new connections between neurons made that can be strengthened, or weakened or pruned away. The neurobiological processes underpinning both cognitive development and functioning such as neurogenesis, synaptic remodelling and neurotransmission, are energetically costly and rely on an uninterrupted supply of mitochondrial adenosine triphosphate (ATP) [
1].
With an average human cortical neuron consuming 4.7 billion ATP molecules per second for ‘housekeeping’ activities and maintenance of its membrane potential and ion homeostasis, mitochondria must produce a staggering 5.7 kg of ATPs each day just to support basic brain function (that’s 5 times the brain’s own weight!) [
2]. Therefore, even though the brain accounts for just 2% of the average human body mass, it’s the most energetically taxing, utilising 25% of total energy supplies [
3]. Moreover, 70–80% of that energy is used by neurons [
2] and primarily at synapses, where mitochondria are largely localized and needed to support the energetic expenditure associated with information processing and propagation of electrical signals [
4].
While the unproportionally high energy requirement of neurons has almost certainly played a central role in the development of human’s uniquely superior cognitive abilities, it has also rendered the brain highly vulnerable to perturbances in energy supply. Not surprisingly, mitochondrial disorders, characterised by bioenergetic failure resulting from mutations in nuclear and/or mitochondrial genes, predominantly affect the brain and commonly cause cognitive impairments ranging from mild to severely debilitating [
5]. Even physiologically healthy aging is associated with reduced availability of glucose for mitochondrial phosphorylation, diminished activity of electron transport chain, deficient antioxidant capacity, and breakdown of mitochondrial energetic function. All these processes are marked by morphological disturbances and alterations in genes regulating mitochondrial biogenesis, which correlate with cognitive decline occurring in later life.
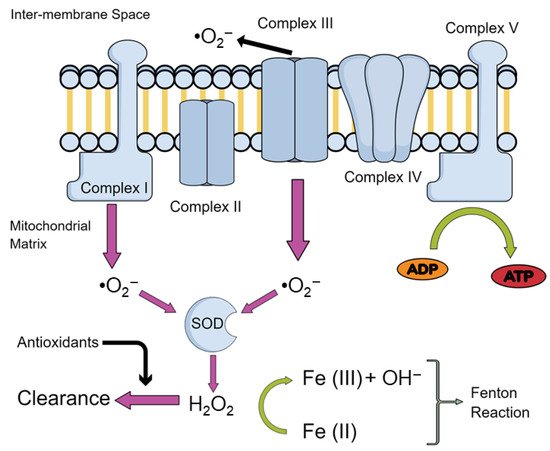
Therefore, the study of mitochondrial impairments is of growing interest in order to unravel the mechanism leading to normal ageing. One of the more prominent theories revolves around the production of reactive oxygen species (ROS). The mitochondrial theory of ageing postulates that production of these ROS derive mainly from oxidative phosphorylation (OXPHOS) taking place within mitochondria. An inevitable by-product of electron transport chain (ETC) activity, responsible for OXPHOS, is the formation of superoxide anion radicals (O
2−), mostly by complexes I and III [
6] and hydroxyl (OH
−) via iron-mediated reduction, known as the Fenton reaction [
7]—overviewed in .
Figure 1. Production of ROS in Mitochondria. An overview of the production of the main reactive oxygen species (ROS) in the mitochondria. Abbreviations: Superoxide ions, •O2−; iron ions, Fe (II)/(III); adenosine diphosphate, ADP; adenosine triphosphate, ATP; superoxide dismutase, SOD; hydrogen peroxide, H2O2.
Under physiological conditions, ROS are involved in processes such as immune response, inflammation, as well as synaptic plasticity, learning, and memory [
8,
9]. However, when produced in excess, ROS production may overwhelm antioxidant defences, leading to impairments of cellular function such as damaging proteins and deoxyribonucleic acid (DNA), and inducing lipid peroxidation which leads to mitochondrial dysfunction observed in aging [
10].
Diet has emerged as a critical component underlying aging-related cognitive deficits. It has been proven that unhealthy diets have an effect on cognition through the induction of an inflammatory response in the aged brain [
11,
12,
13,
14]. Given the prevalence of neuroinflammation in memory impairments, highly prevalent in aged individuals, lifestyle modifications with special emphasis on diet present a promising potential intervention [
15,
16,
17]. Indeed, adherence to the Mediterranean diet [
18] has been associated with better cognitive function [
19,
20] and slower cognitive decline [
19,
20,
21], though some studies have reported null findings [
22,
23,
24].
2. ROS and Its Effects on the Mitochondria
As we have previously described, the imbalance between free radical production and detoxification leads to bioenergetic impairments as well as disturbances in the reduction-oxidation (redox) homeostasis in the brain with increasing age. Indeed, post-mortem brain samples from 80-year-old subjects have revealed a progressive age-related rise in protein nitration and oxidation, together with a decrease in antioxidant defences such as superoxidase dismutase (SOD), catalase (Cat), and glutathione (GSH) reductase activity, as well as mitochondrial complex I activity, mainly in the hippocampus and frontal cortex [
25]. Moreover, Mandal et al. (2012) [
26] have found a gradual decrease in GSH content in the human brain of 56-years-old subjects. More interestingly, this age-related increase in brain oxidative stress was even greater in individuals with high body mass index and in smokers, highlighting the influence of lifestyle in the processes of aging. In line with these findings, Rebrin et al. (2007) [
27] found a decrease in the reduced/oxidized glutathione (GSH/GSSG) ratio in the cortex, striatum and the hippocampus of mice, which are brain regions linked to age-related loss of cognitive function.
During aging, a mitochondrial depolarization process and uncoupling of the OXPHOS have been found due to the aperture of the mitochondrial permeability transition pore (mPTP) [
28]. The mPTP allows free movement of molecules into the mitochondria that weigh less than 1.5 kilo-Dalton. Whilst under healthy conditions this is hypothesised to act as an immediate resource dump of ions such as calcium, prolonged opening is associated with pathological conditions [
29]. ROS may activate the mPTP, which results in the activation of an apoptotic pathway via the release of cytochrome c oxidase and initiation of the caspase-9 cascade [
30]. mPTP activation may also result in further production of ROS as well as in the activation of the nod-like receptor pyrin domain 3 (NLRP3) inflammasome [
31]. This latter finding is relevant since the activation of NLRP3 in microglia and astrocytes in the brain are crucial for neuronal loss in nigrostriatal neurons and remains a key mechanism through which motor deficits and cognitive deficits are manifested in Parkinson’s disease and other disorders [
31,
32,
33]. Moreover, it has been shown that the mPTP is also more susceptible to activation in older mice, thus resulting in increased apoptosis in aged animals [
34]. Furthermore, tumour necrosis factor alpha (TNF-α) has been reported to cause aberrant opening of mPTP, suggesting that agents which increase inflammation with subsequent release of TNF-α, such as high fat diets [
35,
36], may also result in an increase in mPTP. These results suggest that mitochondrial dysfunction, ROS, and inflammatory by-products are responsible for age-related cognitive deterioration and can be further modulated by dietary changes.
2.1. Oxidative Damage to Lipids and Protein in Aging
Aging has been shown to increase the oxidative damage to DNA, lipids [
37,
38,
39] and proteins [
40,
41]. Among phospholipids, cardiolipin became relevant not only for being almost exclusively located in the inner mitochondrial membrane, where it is biosynthesized [
42,
43,
44], but also because evidence suggested that cardiolipin is involved in the regulation of key mitochondrial inner membrane proteins’ activity involved in oxidative phosphorylation [
38,
39,
45]. Additionally, due to its location near the site of ROS production, cardiolipin is particularly prone to be peroxidised. Indeed, oxidative stress readily decreases cardiolipin, which results in a decrease in cytochrome c oxidase, a key enzyme in the ETC [
46,
47]. These results point out that the oxidation/depletion of cardiolipin with aging contribute to mitochondrial dysfunction, leading to cellular dysfunction and eventually cell death.
Lipid peroxides may themselves have a pathogenic role by attacking amino acid residues in proteins such as cytochrome c oxidase, ATPase, and nicotinamide adenine dinucleotide hydrogen (NADH) dehydrogenase, that are prone to oxidation [
48,
49,
50,
51,
52]. This results in heterogenous chemical bonding linking both the protein and lipid together causing peroxidative damage due to loss of solubility [
53]. Peroxidative damage can reduce mitochondrial cytochrome c oxidase activity, but may have more widespread implications through peroxidation of synaptic proteins, which impairs cognition [
54,
55,
56]. Protein oxidation may also occur due to direct interaction with ROS through redox reactions. Additionally, superoxide ions can cause iron-sulphur centres in complex I of the ETC to expel iron, thereby increasing the concentration of labile iron molecules [
57]. Iron is necessary for the reduction of hydrogen peroxide to hydroxide ions via the Fenton reaction [
7], with an increase in iron concentration being linked to ROS production as well as cellular death and aging [
58].
2.2. Lipofuscinosis as a Mitochondrial Dysfunction Marker in Aging
Interestingly, iron accumulates in a heterogenous aggregate known as lipofuscin. Lipofuscin is known as the wear and tear pigment, containing indigestible proteins, lipids and metals, and positively correlates with aging and oxidative stress [
59,
60,
61,
62,
63,
64,
65,
66,
67]. Despite being a well described feature of aging, the exact mechanism through which these macromolecules accumulate remains unknown, as well as any function or deleterious effect they may pose to the cell [
64]. For example, increased intracellular concentrations of iron increase ROS, but it is not known whether sequestered iron in lipofuscin is catalytically active in forming hydroxide ions. Lipofuscin may act as a protective accumulate in this instance by removing the cell’s potential to create ROS. Moreover, a large constituent of lipofuscin is a subunit c of mitochondrial ATP Synthase (SCMAS), the final complex in the ETC [
68]. Why this particular subunit accumulates remains unexplored, however, it is hypothesised that SCMAS may be required in the formation of the mPTP. Therefore, perhaps accumulations of SCMAS reflect an equal increase in mPTP. This might indicate early mitochondrial dysfunction in diseases such as the neuronal ceroid lipofuscinoses as well as lysosomal disruption [
69,
70]. Moreover, homologs in
C. elegans have suggested that removal of this protein reduces mitochondrial function as well as reducing lipofuscin aggregates. Despite the reduction in mitochondrial function, lifespan was extended which may suggest that mPTP activation and cell death is increased when SCMAS is present. Alternatively, SCMAS and lipofuscin aggregates may have a different pathogenic role, distinct from the mPTP, in cell death that remains unexplored [
71]. Therefore, the interplay between appropriate SCMAS levels to enable proper mitochondrial function and keeping lipofuscin to a minimum may be key to delaying mitochondrial dysfunction in aging.
2.3. Oxidative Damage to DNA in Aging
Particularly relevant to aging is the oxidative damage to DNA, specifically mitochondrial DNA (mtDNA) which is localised close to where the production of ROS occurs. Damage to mtDNA persists longer and repairs less easily than nuclear DNA, making it particularly vulnerable to perturbation [
72]. mtDNA only contains one non-coding region and therefore makes mutations in exonic regions very high—likely to give rise to frameshift mutations or deletions. Lesions to mitochondrial DNA such 7,8-dihydro-8-oxo-deoxyguanosine (8-oxo-dG) caused by ROS are indicative of the extent of oxidative stress, and its presence has been noted in mtDNA of aged animals [
73,
74,
75,
76]. As a consequence, DNA repair mechanisms to 8-oxo-dG lesions via mitochondrial apurinic/apyridinimic endonucleases are upregulated in aged rats compared to younger rats [
77]. Whilst the same study reported an increase in the DNA glycosylase repair mechanism, another study has shown reductions of DNA glycosylase in old mice, making it difficult to draw conclusions [
78]. Indeed, the vast majority of DNA repair mechanisms reduce in activity with age, so it is possible that is a very specific increase to 8-oxo-dG lesions [
79,
80].
Accumulation of mtDNA mutations via ROS has been suggested as a central mechanism driving aging and age-related diseases [
81,
82,
83,
84,
85,
86] due to their contribution to cellular senescence [
87], a process which halts cell division and thereby prevents new cell formation. The cause of senescence is believed to be the shortening of telomeres, which are the caps that protect chromosomes from damage response enzymes [
88]. Telomere shortening is directly related to oxidative stress levels with ROS, able to manipulate telomere maintenance through multiple pathways [
89,
90]. ROS-induced 8-oxo-dG lesions in mtDNA have been shown to reduce the ability for telomerase to bind to telomeres as well as reduce the activity of the reverse transcriptase subunit of telomerase [
91,
92]. Telomerase is critical for the extension of telomere length due to its ability to add guanine-rich repeats and increase the length of the chromosome. Antioxidants have been shown to increase the activity of the reverse transcriptase subunit and delay cellular senescence [
92]. Furthermore, female rats fed a telomerase activator-65 (TA-65, which increases telomerase activity), purified from the root of Chinese herbs such as Astragalus membranaceus, after a brain injury showed increased cognitive function, movement, and reductions in depressive-like behaviours [
93]. Other studies support these findings and also show telomerase-dependent neurogenesis and an increased lifespan, as well as the ability to supplement TA-65 through diet [
94] or increase telomerase directly through activities such as meditation [
95], which may be able to delay the onset of cellular senescence [
94,
96].
In addition to telomerase binding, 8-oxo-dG also reduces TTAGGG repeat binding factor 1 (TRF1) and 2 (TRF2) binding affinities. These proteins are components of the telomere cap, thus the presence of mtDNA lesions renders chromosomes more vulnerable to DNA damage response enzymes [
97,
98,
99]. ROS builds upon this vulnerability by also upregulating the DNA-damage response, which increases levels of p53 [
100,
101]. P53 then positively feeds back to regulate telomeric capping by ubiquitination and degradation of TRF2, thereby destabilizing the telomere caps and causing early onset senescence [
102,
103].
This entry is adapted from the peer-reviewed paper 10.3390/ijms22073574