Nitroaromatic antibiotics show activity against anaerobic bacteria and parasites, finding use in the treatment of Heliobacter pylori infections, tuberculosis, trichomoniasis, human African trypanosomiasis, Chagas disease and leishmaniasis.
- nitroaromatic antibiotics
- nitric oxide (NO)
- nitrite
- nitroxyl (HNO)
1. Introduction
Nitroaromatic compounds contain one or more nitro groups directly bonded to an aromatic group (Figure 1) [1].
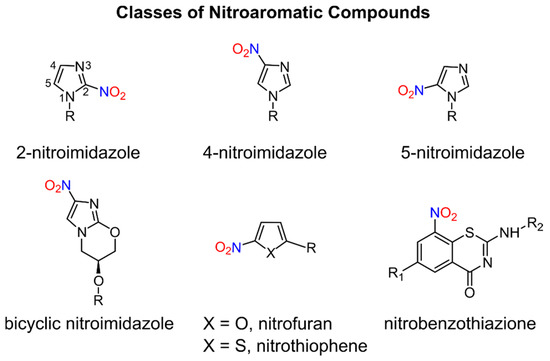
Figure 1. General classes of biologically active nitroaromatic compounds.
Numerous nitro heteroaromatic compounds, especially those containing imidazole and furan or thiophene groups as depicted in Figure 1, demonstrate biological activities allowing for their clinical use as antibiotics and are discussed in this review.
The chemical and physical properties of the nitro group (−NO2) including its electron-withdrawing ability, polarity, size, ability to form hydrogen bonds and redox properties [1][2] contribute to its key role in the action of many drugs, especially antimicrobial agents. Nitroaromatic compounds find use in the treatment of a wide variety of infectious diseases for both bacterial infections, including tuberculosis (TB), and parasitic infections such as giardiasis, trichomoniasis, human African trypanosomiasis (HAT), Chagas disease and leishmaniasis, which are increasingly becoming global health threats [3]. Despite the antibacterial and antiparasitic properties of nitroaromatic antibiotics, drugs containing nitro groups often demonstrate mutagenicity and unacceptable toxicity profiles, which have hindered further development of this drug class. The potent biological activity of these compounds drives the search for non-mutagenic and selectively toxic nitroaromatic antibiotics that kill the infectious agent without damaging host cells [1][4]. In addition, repurposing existing nitroaromatic antibiotics that have been proven safe represents a less risky and cost-efficient strategy in the search of new and effective drugs to treat neglected tropical diseases (NTD) [3].
The redox biochemistry of the nitro group plays a central role in the biological activity of nitroaromatic antibiotics. These compounds require reductive bioactivation for antimicrobial activity with the reduction being carried out by nitroreductases (NTRs) of type 1 or 2 (or type I or II, Scheme 1) that possess reducing power greater than the reduction potential of the nitroaromatic compound. Numerous organism-specific nitroreductases exist as well as other systems including pyruvate/ferredoxin oxidoreductase and hydrogenases that are capable of catalyzing nitro group reduction [5][6][7][8][9][10].
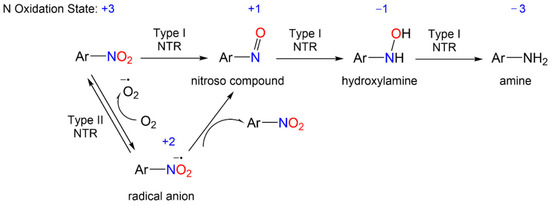
Scheme 1. General reduction mechanism of nitroaromatic compounds.
2. Nitrofurans/Nitrothiophenes
A number of nitro-substituted furans and thiophenes have a long history of use as treatments of anaerobic bacterial infections and parasitic diseases (Figure 2). As with other nitro-substituted aromatic therapeutics, biological activity requires metabolic bioreduction. Early work shows NO2− release upon exposure to GSH and liver extracts from some 2-substituted 5-nitrofurans, and NO generation upon chemical reduction of nitro furans has also been reported [11][12]. Representative examples discussed below include nifurtimox, nitrofurantoin and molecules of the nitrothiophene and nitrothienopyrimidine classes (Figure 2).
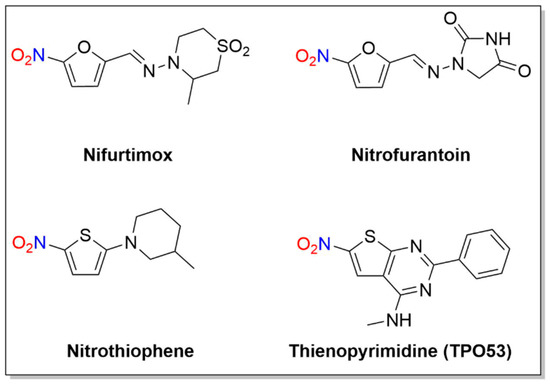
Figure 2. Structures of nifurtimox, nitrofurantoin and thienopyrimidine.
2.1. Nifurtimox
Nifurtimox, first introduced in 1965 [13], is a nitrofuran with activity against trypomastigotes and amastigotes and is used to treat Chagas disease, as well as HAT (Figure 2) [13].
Nifurtimox acts as a prodrug after absorption from the gastrointestinal tract and liver metabolism, where nitroreduction is catalyzed by cytochrome P450 reductase and NTR2, leading to oxidative stress, the generally accepted trypanocidal mechanism of action for nifurtimox [14]. More specifically, nifurtimox metabolism by a trypanosomal NTR1 proceeds through the nitroso and hydroxylamine intermediates, resulting in generation of a cytotoxic ring-opened unsaturated nitrile metabolite as determined by LC-MS (Scheme 2) [14].
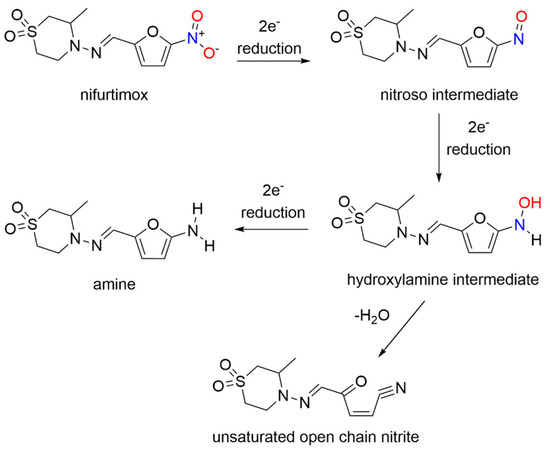
Unlike nifurtimox, this metabolite inhibits both parasite and mammalian cell growth. This study sheds light on the selective toxicity of nifurtimox as a result of NTR1 activation, which generates secondary toxic organic products rather than nitrogen oxides.
2.2. Nitrofurantoin
Nitrofurantoin, first patented in 1952 [15], is an antibiotic used to prevent and treat bacterial urinary tract infections (e.g., E. Coli, Enterobacter cystitis, Enterococcus, Klebsiella and Staphylococcus aureus) (Figure 2). The mechanism of action of nitrofurantoin is complex and includes bacterial nitroreductase-catalyzed formation of various reactive intermediates [16]. While reduction of nitrofurantoin generates nitro radical anions in mammalian cells [17], speculation exists whether NO is produced as other early studies found that enzymatic reduction of nitrofurantoin generates ROS [18].
The relationship of NO release and the biological activity of nitrofurantoin was examined on intracellular uropathogenic E. coli (UPEC), which causes urinary tract infections (UTI) [19]. Both extracellular and intracellular NO production of infected 5637 bladder cells treated with either the NO donor (2,2′-(hydroxynitrosohydrazono)bis-ethanimine (DETA)/NO)) or nitrofurantoin were measured through either the Griess assay in combination with UV–VIS spectrometry, or DAF-FM-DA and confocal microscopy [19]. While nitrofurantoin was effective against colonized UPEC, neither NO2− nor NO was detected in the infected 5637 bladder cells treated with nitrofurantoin through either measurement, indicating that NO is not a key byproduct in the mammalian metabolism of nitrofurantoin [19].
The cytotoxicity of nitrofurantoin in the dark and after light exposure (UVA irradiation, wavelength = 385 nm) was studied in murine melanoma B16F10 cells for a role for NO [20]. Optical absorption and fluorescence spectroscopies, chemiluminescence, fluorescence image microscopy and standard cell viability tests detected NO and its effect on the cells during nitrofurantoin irradiation in both buffered solution and cells [20]. The mechanism of cell death in the dark results from enzymatic NO release, while light exposure increases nitrofurantoin cytotoxicity through increased NO photorelease [20]. The nitrofurantoin cytotoxicity was several times greater upon light exposure [20], which could render this drug a promising photochemotherapy option.
2.3. Nitrothiophenes/Thienopyrimidine (TP053)
Given the similarity of thiophene to furan and imidazole ring systems, a series of 5-nitrothiophenes was evaluated against MTB, revealing a highly active yet non-toxic or mutagenic lead compound (2-(3-methylpiperidin-1-yl)-5-nitrothiophene) from these assays (Figure 2) [21]. Mechanistic studies indicate this 5-nitrothiophene requires the F420-dependent nitroreductase (Ddn) for activity and these compounds generate NO2− during incubation with recombinant M. smegmatis containing Ddn [21]. The presence of a nitro group at the C-5 position of the thiophene ring is critical for activity as exchange of the nitro group to an acetyl group or movement of the nitro group from the C-5 to the C-3 position abolishes activity [21]. These compounds appear to act in a nearly identical fashion as the bicyclic 4-nitroimidazoles (e.g., PA-824 and OPC-67683) and their ability to kill MTB and release NO2− indicates these thiophene heterocycles can release nitrogen oxides in a similar fashion as the more complicated bicyclic nitroimidazoles, identifying new lead therapeutic compounds [21].
The mechanism of action was studied for the anti-tubercular thienonpyrimidine prodrug (TP053), a fused thiophene–pyrimidine, active against both replicating and non-replicating MTB [22]. Activation of TP053 by a mycothiol-dependent nitroreductase (Mrx2) releases NO and/or NO2− as judged by the Griess analysis, similar to PA-824 activation by Ddn [22]. Based on previous studies of 2-nitrofuran reduction [12], a suggested mechanism for NO release involves the conversion of the nitro group (-NO2) to a nitrite ester (-ONO) that apparently homolyzes to NO [22]. Further reduction and dearomatization of the thiophene ring gives a thiophenol derivative, as confirmed by LC-MS [22]. This thiophenol (e.g., 2-(4-mercapto-6-(methylamino)-2-phenylpyrimidin-5-yl)ethan-1-ol) contributes to the antimycobacterial effects on both replicating cells and non-replicating cells [22]. These results identify yet another nitroaromatic substrate with a different reducing system capable of producing nitrogen oxides.
2.4. 1,3-Benzothiazin-4-ones (BTZs)
BTZ043
Similar to PA-824 and its use as an anti-TB agent, 1,3-benzothiazin-4-ones (BTZs, especially BTZ043) and related nitroaromatic compounds in the nitrobenzothiazione class have emerged as effective candidates for the same purpose (Figure 3) [23].
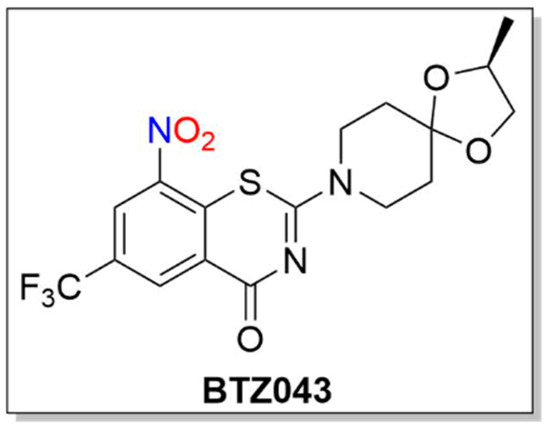
Figure 3. Structure of BTZ043.
BTZs kill MTB in vitro, ex vivo and in mouse models of TB [24]. Based on potency and specificity for mycobacteria, BTZ0838 (2-[2-methyl1,4-dioxa-8-azaspiro[4.5]dec-8-yl]-8-nitro-6-(trifluoromethyl)-4H-1,3-benzothiazin-4-one)) was synthesized in seven steps, and its enantiomers BTZ043 (S) and BTZ044 (R) were evaluated in a structure function study [24]. Both the sulfur atom and the nitro group are necessary for activity as chemical reduction of the nitro group to the hydroxylamine/amine diminishes activity. Nitroreductase-producing mycobacteria were resistant to BTZ043 [24]. BTZ043 acts as a prodrug and functions through nitro group reduction to the nitroso, hydroxylamine and ultimately the amine [25]. Both genetic and biochemical studies identify decaprenylphosphoryl-beta-D-ribose 2′-epimerase (DprE) as the target of BTZ [25]. Suicide inhibition of DprE by BTZ043 prevents the formation of decaprenylphosphoryl arabinose, a crucial precursor needed for cell wall arabinan synthesis, leading to death of the bacteria [25]. BTZ043 and nitrobenzamide hybrids react with cysteine thiolates or hydrides at the active site of DprE, inducing the nonenzymatic reduction of the nitro group of BTZ043 to the nitroso intermediate through nucleophilic addition at the unsubstituted electron-deficient aromatic carbon present in these compounds, which leads to inhibition of DprE1 (Scheme 3) [23].
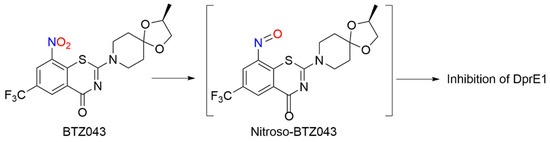
Scheme 3. Reduction of BTZ043 [23].
This reactivity mirrors the classical von Richter reaction and provides an alternative hypothesis for the mechanism of action for nitroaromatic anti-TB agents, such as BTZ043 [23]. While evidence of NO formation from BTZ043 does not exist or is suggested, these results reveal the rich bio-organic chemistry of these compounds specifically and nitroaromatic antibiotics in general [23]. Based on the promise of BTZs as a novel class of TB drug candidates, a ligand-based design model, based on MIC values from BTZ [24] and predictive solubility, was used to yield a variety of compounds of greater activity than BTZ043 against M. smegmatis and MDR-TB with increased solubility, as well as microsomal, metabolic and plasma stability, and low toxicity [26].
This entry is adapted from the peer-reviewed paper 10.3390/biom11020267
References
- Patterson, S.; Wyllie, S. Nitro Drugs for the treatment of trypanosomatid diseases: Past, present, and future prospects. Trends Parasitol. 2014, 30, 289–298.
- Nepali, K.; Lee, H.Y.; Liou, J.P. Nitro-group-containing drugs. J. Med. Chem. 2019, 62, 2851–2893.
- Ang, C.W.; Jarrad, A.M.; Cooper, M.A.; Blaskovich, M.A.T. Nitroimidazoles: Molecular fireworks that combat a broad spectrum of infectious diseases. J. Med. Chem. 2017, 60, 7636–7657.
- Strauss, M.J. The Nitroaromatic group in drug design. pharmacology and toxicology (for nonpharmacologists). Ind. Eng. Chem. Prod. Res. Dev. 1979, 18, 158–166.
- Martínez-Júlvez, M.; Rojas, A.L.; Olekhnovich, I.; Angarica, V.E.; Hoffman, P.S.; Sancho, J. Structure of RdxA-an oxygen-insensitive nitroreductase essential for metronidazole activation in Helicobacter pylori. FEBS J. 2012, 279, 4306–4317.
- Race, P.R.; Lovering, A.L.; White, S.A.; Grove, J.I.; Searle, P.F.; Wrighton, C.W.; Hyde, E.I. Kinetic and structural characterisation of Escherichia Coli nitroreductase mutants showing improved efficacy for the prodrug substrate CB1954. J. Mol. Biol. 2007, 368, 481–492.
- Hall, B.S.; Wu, X.; Hu, L.; Wilkinson, S.R. Exploiting the drug-activating properties of a novel trypanosomal nitroreductase. Antimicrob. Agents Chemother. 2010, 54, 1193–1199.
- Searle, P.F.; Chen, M.J.; Hu, L.; Race, P.R.; Lovering, A.L.; Grove, J.I.; Guise, C.; Jaberipour, M.; James, N.D.; Mautner, V.; et al. Nitroreductase: A prodrug-activating enzyme for cancer gene therapy. Clin. Exp. Pharm. Physiol. 2004, 31, 811–816.
- Müller, J.; Rout, S.; Leitsch, D.; Vaithilingam, J.; Hehl, A.; Müller, N. Comparative characterisation of two nitroreductases from giardia lamblia as potential activators of nitro compounds. Int. J. Parasitol. Drugs Drug Resist. 2015, 5, 37–43.
- Bryant, C.; DeLuca, M. Purification and characterization of an oxygen-insensitive NAD(P)H nitroreductase from Enterobacter Cloacae. J. Biol. Chem. 1991, 266, 4119–4125.
- Boyland, E.; Speyer, B.E. Enzyme-catalysed reactions between some 2-substituted 5-nitrofuran derivatives and glutathione. Biochem. J. 1970, 119, 463–472.
- Grigor, N.B.; Chechekin, G.V.; Arzamaztsev, A.P.; Levina, V.I.; Granik, V.G. Nitrogen oxide generation during reduction of nitrofuran antibacterial drugs. Chem. Heterocycl. Compd. 1999, 35, 788–791.
- Bern, C.; Montgomery, S.P.; Herwaldt, B.L.; Rassi, A.; Marin-Neto, J.A.; Dantas, R.O.; Maguire, J.H.; Acquatella, H.; Morillo, C.; Kirchhoff, L.V.; et al. Evaluation and treatment of chagas disease in the United States: A systematic review. J. Am. Med. Assoc. 2007, 298, 2171–2181.
- Hall, B.S.; Bot, C.; Wilkinson, S.R. Nifurtimox activation by trypanosomal Type I nitroreductases generates cytotoxic nitrile metabolites. J. Biol. Chem. 2011, 286, 13088–13095.
- Arredondo-Garcı, L.; Amabile-Cuevas, C.F. Nitrofurantoin, phenazopyridine, and the superoxide-response regulon SoxRS of Escherichia coli. J. Infect. Chemother. 2013, 19, 1135–1140.
- McOsker, C.C.; Fitzpatrick, P.M. Nitrofurantoin: Mechanism of action and implications for resistance development in common uropathogens. J. Antimicrob. Chemother. 1994, 33, 22–30.
- Ramakrishna Rao, D.N.; Mason, R.P. Generation of nitro radical anions of some 5-nitrofurane, 2- and 5-nitroimidazoles by norepinephrine, dopamine, and serotonin. A possible mechanism for neurotoxicity caused by nitroheterocyclic drugs. J. Biol. Chem. 1987, 262, 11731–11736.
- Youngman, R.J.; Osswald, W.F.; Elstner, E.F. Mechanisms of oxygen activation by nitrofurantoin and relevance to its toxicity. Biochem. Pharm. 1982, 31, 3723–3729.
- Vumma, R.; Bang, C.S.; Kruse, R.; Johansson, K.; Persson, K. Antibacterial effects of nitric oxide on uropathogenic escherichia coli during bladder epithelial cell colonization-a comparison with nitrofurantoin. J. Antibiot. 2016, 69, 183–186.
- Ferreira, L.P.; Parra, G.G.; Codognato, D.C.K.; Amado, A.M.; Da Silva, R.S. Light induced cytotoxicity of nitrofurantoin toward murine melanoma. Photochem. Photobiol. Sci. 2017, 16, 1071–1078.
- Hartkoorn, R.C.; Ryabova, O.B.; Chiarelli, L.R.; Riccardi, G.V.; Makarov, V.; Makarov, S.T. Mechanism of Action of 5-nitrothiophenes against mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 2944–2947.
- Chiarelli, L.R.; Salina, E.G.; Mori, G.; Azhikina, T.; Riabova, O.; Lepioshkin, A.; Grigorov, A.; Forbak, M.; Madacki, J.; Orena, B.S.; et al. New insights into the mechanism of action of the thienopyrimidine antitubercular prodrug TP053. ACS Infect. Dis. 2020, 6, 313–323.
- Tiwari, R.; Moraski, G.C.; Krchňák, V.; Miller, P.A.; Colon-Martinez, M.; Herrero, E.; Oliver, A.G.; Miller, M.J. Thiolates chemically induce redox activation of btz043 and related potent nitroaromatic anti-tuberculosis agents. J. Am. Chem. Soc. 2013, 135, 3539–3549.
- Makarov, V.; Manina, G.; Mikusova, K.; Möllmann, U.; Ryabova, O.; Saint-Joanis, B.; Dhar, N.; Pasca, M.R.; Buroni, S.; Lucarelli, A.P.; et al. Benzothiazinones kill mycobacterium tuberculosis by blocking arabinan synthesis. Science 2009, 324, 801–804.
- Trefzer, C.; Rengifo-Gonzalez, M.; Hinner, M.J.; Schneider, P.; Makarov, V.; Cole, S.T.; Johnsson, K. Benzothiazinones: Prodrugs That covalently modify the decaprenylphosphoryl- β-D-ribose 2′-epimerase DprE1 of mycobacterium tuberculosis. J. Am. Chem. Soc. 2010, 132, 13663–13665.
- Karoli, T.; Becker, B.; Zuegg, J.; Möllmann, U.; Ramu, S.; Huang, J.X.; Cooper, M.A. Identification of antitubercular benzothiazinone compounds by ligand-based design. J. Med. Chem. 2012, 55, 7940–7944.