Inflammatory bowel diseases (IBD) are chronic conditions that primarily affect the gastrointestinal tract, with a complex pathogenesis; they are characterized by a significant heterogeneity of clinical presentations and of inflammatory pathways that sustain intestinal damage. After the introduction of the first biological therapies, the pipeline of therapies for IBD has been constantly expanding, and a significant number of new molecules is expected in the next few years.
- inflammatory bowel disease
- biological therapy
- predictors
- biomarkers
1. Introduction
Crohn’s disease (CD) and ulcerative colitis (UC) represent the two main forms of inflammatory bowel diseases (IBD). These are chronic conditions with a relapsing-remitting course that affect more than 5 million people worldwide, mostly in Western countries; their epidemiology is rapidly changing, with a sharp increase in incidence in Western countries registered in the previous decades [1]. IBD carries a significant direct and indirect health-care burden, which is mainly represented by drugs like biologics and small molecules, but also includes hospitalization-related costs and days of absence from work due to the disease [2]. In this scenario, there is an urgent need for a more effective approach than today’s trial and error method, when it comes to starting therapies.
To date, IBD treatment is based on corticosteroids (for the acute phases), mesalamine (only for UC), traditional immunosuppressants and targeted therapies; this last category includes: anti-tumor necrosis factor α (TNFα), anti-integrin, anti-interleukin (IL) 12/23 and Janus kinases (JAK) inhibitors [3][4][5]. It has been shown that different pathogenic pathways can sustain bowel damage in IBD [6][7], so that two patients with similar clinical phenotypes can have different inflammatory pathways activated and, thus, respond to different targeted therapies. There is also evidence that, within the same patient, the immune system can exhibit a significant plasticity and change the inflammatory pathways that are activated during the course of disease [8]. Such a complexity can easily explain why current therapies are only of limited efficacy. While the armamentarium for IBD treatment is constantly expanding—with new drugs targeting different pathogenetic pathways—there is still a significant proportion of patients who do not respond to therapy: data from clinical trials and real life report clinical efficacy of a single drug in up to 60% of patients [9][10]. Whether these patients would respond to another agent is not possible to foretell, but there is strong evidence that second- and third-line agents can be effective even in the case of primary failure, although to a lesser extent [11]. Furthermore, a substantial percentage of patients experience secondary failures [12]: in these cases, unless surgery is made mandatory by disease complications, the usual choice is to try another medical treatment.
Taken together, these considerations point to the necessity for the development of new prognostic tools able to identify those patients who would benefit from an early introduction of advanced therapies, to predict their response to a specific therapy and to assess response at an early point in treatment. Some clinical features have been identified so far, but they mainly identify patients who would most benefit from immunosuppressive therapies and/or patients who are more likely to respond to medical treatment, while they do not offer much information about patients’ response to a specific drug. Implementation of personalized medicine into IBD routine management represents one of the most compelling challenges of coming years, in order to provide patients with better clinical care in parallel with a reduction of costs for the health-care systems (Figure 1).
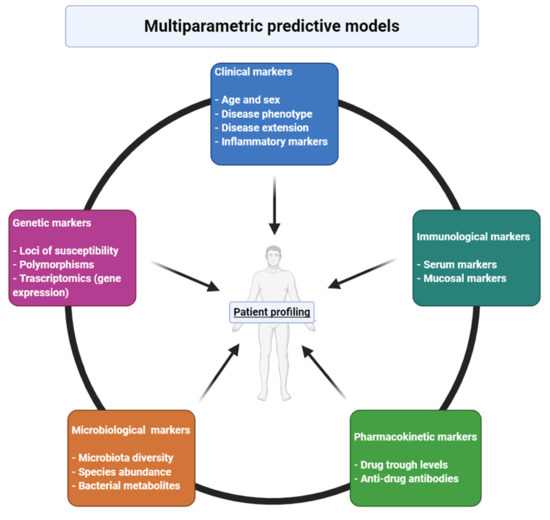
Figure 1. Different features that contribute to responsiveness to a certain drug and that needs to be included in multiparametric models for prediction of response in inflammatory bowel diseases. Created with BioRender.com.
2. Traditional Markers
Associations of patients’ and disease characteristics with response to therapy have been widely investigated, but results have been generally discouraging. Age, gender, weight and smoking status have not been confirmed to correlate with response to anti-TNF or other targeted agents [13]. Two recent meta-analysis suggested that early treatment of CD is associated with better response rates [14][15], and one of those also observed an association with higher rates of mucosal healing [15]; however, no differences between different drugs have been observed so far. In CD patients, disease location has not been found to be associated with treatment outcomes of anti-TNF and ustekinumab [13]; of note, one study reported an association between colonic localization and better responses to vedolizumab [16], that was not confirmed in others. Some studies have reported a correlation, in CD patients, between inflammatory phenotype and better response rates with TNF antagonists, compared to stricturing or penetrating diseases [17][18][19][20]; such findings have not been confirmed for vedolizumab and ustekinumab.
Various serological biomarkers have been investigated for their potential predictive role. In a recent retrospective study on elderly (>60 years old) patients, a higher serum triiodothyronine-to-thyroxine (T3/T4) ratio was found to be associated to mucosal healing, regardless of the biological drug used [21]. C-reactive protein (CRP) levels haves been inconsistently associated with response to biological therapies. A correlation between higher CRP levels and better response to TNF antagonists has been reported in CD patients [22][23][24][25]; conversely, a negative correlation between CRP levels and response rates to TNF inhibitors has been observed in UC [26][27][28]. Similar findings have also been observed with vedolizumab, but not with ustekinumab [13]. However, such results have not been confirmed by many other studies. Higher CRP levels can help identify those patients whose symptoms are actually dependent on active IBD and, in CD, can help discriminate inflammatory vs. stricturing phenotypes; on the other hand, such high levels can also be expression of a higher inflammatory burden, that is comprehensively associated with poorer response to medical treatment. Faecal calprotectin has been tested as a potential predictor of response, with disappointing results. Of note, in a prospective observational study it has been found that lower post-induction calprotectin levels were able to predict sustained clinical response and mucosal healing in IBD patients receiving anti-TNF treatment [29].
Previous exposure to biologics has been associated with poorer response to subsequent lines of therapies. Reason for discontinuation seems to have an impact on the likelihood of responding to second-line therapies: a Spanish retrospective study and 2 meta-analysis concluded that discontinuation due to anti-TNF intolerance was associated with higher rates of response to both second anti-TNF or other biologic agents [11][30][31]. Primary non-response to TNF antagonists seems to correlate with an even lower likelihood of response to second-line therapies, when compared to secondary loss of response (LOR) [30]. A retrospective study on UC patients showed that, in case of primary failure, out-of-class swap seems to be superior to in-class switch with regard to rates of clinical response and remission [32]. However, such findings have not been consistently confirmed in literature.
3. Genetic Markers
More than 240 susceptibility loci for IBD have been identified so far [33]. Such genes have greatly contributed to the understanding of IBD pathogenesis and to the identification of novel therapeutic targets. However, genetic markers have usually performed quite poorly in predicting response to a specific drug [34]. In Table 1, there is an overview of the studies investigating the association between genetic markers and response to therapy.
Table 1. Genetic markers.
|
Study |
Genetic Markers |
Outcomes |
|---|---|---|
|
Bek et al. 2016 [34] |
Polymorphisms in TLR2, rs11938228, TLR4, TLR9, TNFRSF1A, IFNG, IL6 and IL1B (rs4848306) |
Clinical response to anti-TNF in IBD patients |
|
Tong et al. 2013 [35] |
Polymorphisms in TNF-α promoter (-308 A/G and -857 C/T) |
Clinical response to anti-TNF in IBD e SpA patient |
|
Bank et al. 2014 [36] |
Polymorphisms implicated in NF-kB pathway: TLR2, TLR4, TLR9, LY96 (MD-2), CD14, MAP3K14 (NIK), TNFA, TNFRSF1A, TNFAIP3(A20), IL1B, IL1RN, IL6, IL17A, IFNG |
Clinical response to anti-TNF in IBD patients |
|
Jürgens et al. 2010 [37] |
Polymorphisms in IL23R |
Early response to infliximab in UC patients |
|
Sazonovs et al. 2020 [38] |
HLA-DQA1*05 |
Development of ADA against infliximab and adalimumab in CD patients |
|
Billiet et al. 2015 [39] |
HLA-DRB1 |
Development of ADA against infliximab in IBD patients |
|
Louis et al. 2004 [40] |
Polymorphism in IgG Fc receptor IIIa |
Development of ADA against infliximab in CD patients |
|
Niess et al. 2012 [41] |
Polymorphisms in NOD2 |
Clinical response to anti-TNF in CD patients |
|
Juanola et al. 2015 [42] |
Polymorphisms in NOD2 |
Loss of response to anti-TNF in CD patients |
|
Schäffler et al. 2018 [43] |
Polymorphisms in NOD2 |
Lower anti-TNF TLs in CD patients |
|
Koder et al. 2015 [44] |
Polymorphisms in ATG16L1 |
Clinical response to adalimumab in CD patients |
|
Hlavaty et al. 2007 [45] |
Polymorphisms in Fas, Fas ligand and Caspase 9 (Apoptotic Pharmacogenetic Index) |
Clinical response to infliximab in CD patients |
|
Barber et al. 2016 [46] |
Multiple polymorphisms (Combined clinical-genetic model) |
Short- and long-term to anti-TNF in CD patients |
|
Burke et al. 2018 [47] |
Multiple polymorphisms (Combined clinical-genetic model) |
Short- and long-term response to anti-TNF in UC patients |
|
Wang et al. 2019 [48] |
Polymorphisms in TNFSF4/18, PLIN2, rs762787, rs9572250, rs144256942, rs523781 |
Clinical response to anti-TNF in IBD patients |
ADA: anti-drug antibodies; ATG16L1: autophagy-related 16 like 1; CD: Crohn’s disease; HLA: human leukocyte antigens; IBD: inflammatory bowel disease; IL: interleukin; MAP3K14: mitogen-activated protein kinase kinase kinase 14; NF-kB: nuclear factor kappa-light-chain-enhancer of activated B cells; NOD2: nucleotide-binding oligomerization domain-containing protein 2; PLIN2: perilipin 2; TLR: toll-like receptor; TNF: tumor necrosis factor; TNFR: tumor necrosis factor receptor; UC: ulcerative colitis; TNFSF: tumor necrosis factor superfamily; IFNG: interferon gamma.
Genome-wide association studies have reported that disease susceptibility loci do not seem to substantially contribute to anti-TNF non-response. For instance, polymorphisms in the genes encoding for TNF or molecules involved in the TNF receptor pathway have been inconsistently associated with treatment response. A 2013 meta-analysis reported an association between 2 polymorphisms in the TNF promoter and responsiveness to TNF inhibition in IBD patients: specifically, the more common alleles were associated with better response rates [35]. Another meta-analysis found a positive correlation between polymorphisms in FCGR3A, TLR4, TNFRSF1A, IFNG, IL6, IL1B genes and better clinical response, whereas variants of TLR2 and TLR9 were negatively correlated [34]. Polymorphism in the Nuclear Factor kappa-light-chain-enhancer of activated B cells (NFkB) pathway, TNF pathway and pathways of other cytokines have been linked to treatment response in IBD patients treated with anti-TNF agents [36][49]. IL23 receptor polymorphisms have been associated with response to infliximab in UC patients [37]. Moreover, the HLA-DQA1*05, the HLA-DRB1 allele and polymorphisms at the FCGR3A locus (encoding IgG Fc receptor IIIa) have been correlated with an increased risk of anti-drug antibodies (ADA) formation in CD patients treated with anti-TNF agents [38][39][40]. Being large complex proteins, monoclonal antibodies—especially infliximab, that is a chimeric antibody—can stimulate the production of ADA, which are associated with treatment inefficacy [50]. Identifying patients who are more likely to develop ADA would be of great help, as we know that concomitant immunosuppression (with thiopurines and methotrexate) reduces the risk of their formation [51].
Another marker previously identified by gene array studies in mucosal biopsies of IBD patients is the IL13 receptor alpha 2 (IL13RA2) [52]. This biomarker has been more recently evaluated as mRNA expression in the mucosa of IBD patients prior to therapy and found to be specifically predictive of non-response to anti-TNF in terms of mucosal healing at 6 months with an area under the receiver operating characteristic (AUROC)of 0.90 for infliximab and 0.94 for adalimumab, p < 0.001 [53].
The NOD2 gene is associated with CD susceptibility and with a more aggressive course of disease [54][55]; it encodes for a protein that plays a role in eliciting the immune response and is implicated in the inflammatory pathway of TNF. Some studies have found an association between NOD2 variants and worse response to anti-TNF therapy [41][42][43]. Polymorphisms in the ATG16L1 gene have been associated better response rates [44] and longer benefit [46] in CD patients treated with TNF antagonists. An apoptotic pharmacogenetic index (API) has been proposed to predict treatment response to anti-TNF in CD patients. The index was based on single nucleotide polymorphisms of 3 apoptotic genes (Fas, Fas-ligand and caspase-9). The authors elaborated a model, combining API with clinical features, that was able to predict response to infliximab in luminal and penetrating CD [45]. Predictive models combining clinical and genetic features have been shown to be superior to models based on clinical characteristics only for predicting primary non-response to anti-TNF agents in CD (Area Under the Receiver Operating Characteristics [AUROC] 0.93 vs. 0.70, p < 0.0001) [46], UC (AUROC 0.87 vs. 0.57, p < 0.0001) vs. [47] and IBD patients (AUROC 0.89 vs. 0.72, p < 0.0001) [48].
Data on genetic variants associated with response to anti-integrin is scarce. In the phase 2 trial of etrolizumab (anti-β7 integrin) in UC patients, αE gene expression was found to be predictive of clinical remission at week 10 [56]. This result was subsequently enhanced by the finding that also higher levels of Granzyme A (which is highly expressed by αE+ cells) mRNA in colonic biopsies taken at baseline could identify UC patients in clinical remission at week 10 of etrolizumab therapy [57]. No association between genetic markers and anti-IL12/23 response has been identified so far.
4. Other Markers: Transcriptomics, Proteomics and Immunological Markers
Transcriptomics studies have provided some more pleasing results, suggesting that therapy response seems to rely on differential expression of significant genes, more than on genetic variants.
A putative biomarker identified by transcriptomics studies is the triggering receptor expressed on myeloid cells 1 (TREM-1), although with some discordant findings: a study of gene expression described a down-regulation in whole blood of non-responders to anti-TNF. However, these patients showed an up-regulation of TREM-1 and of chemokine receptor type 2 (CCR2)–chemokine ligand 7 (CCL7) in intestinal biopsies. In the same study, plasma cell frequencies were examined in intestinal biopsies by CD138+ staining and were considered able to predict anti-TNF response, being higher in non-responders (p = 0.0005). [58] A different study analyzed the expression of TREM1 in whole blood and in mucosal tissue and as protein level in the serum and found a significant reduction in IBD patients who achieved mucosal healing [59]. This pathway seems to be specific for anti-TNF response as no modifications was detected in patients treated with vedolizumab or ustekinumab.
Measures of TNF production have been studied as putative biomarkers of response to anti-TNF. In vivo imaging by endomicroscopy revealed higher numbers of mTNF+ cells in short-term (12 weeks) responders, after local fluorescent adalimumab administration [60]. A recent study analyzed in 42 IBD patients the in vitro production of TNF from peripheral blood mononuclear cells (PBMC) stimulated with lipopolysaccharide (LPS) and found it to be predictive of clinical response after 6 weeks of infliximab therapy. A cutoff of 500 pg/mL was identified in CD for short-term response with 100% sensitivity and 82% specificity [61].
High expression of a member of the IL6 family, oncostatin M (OSM), in the intestinal mucosa was found to predictive of refractoriness to anti-TNF therapy. The clinical response was assessed at week 8 and 30 in a cohort of patients treated with infliximab (from ACT1/2 studies) and at 6 weeks in a cohort of patients treated with golimumab (from the Program of Ulcerative Colitis Research Studies Utilizing an Investigational Treatment−Subcutaneous, PURSUIT study) [62]. This biomarker was also studied at baseline in serum and found to be predictive of mucosal healing at 54 weeks of infliximab treatment with an AUC of 0.91 in a study on 45 CD patients [63].
Assays of α4β7 occupancy in peripheral blood T cells showed almost complete blocking of this integrin in patients after vedolizumab treatment, irrespective of clinical response and also of circulating drug levels [64]. However, in a small study, vedolizumab responders had higher pre-treatment α4β7 expression on T effector memory cells (p = 0.0009 for CD4 and.0043 for CD8) and on natural killer (NK) cells (p = 0.0047) [65]. These results were partly confirmed at the tissue level by a preliminary study with confocal endomicroscopy with fluorescein isothiocyanate (FITC) labelled vedolizumab, where only CD patients were responders to subsequent therapy with the anti-integrin showed α4β7+ cells in the mucosa [66]. A study examined the in vitro assay of baseline peripheral blood CD4+ cells dynamic adhesion to recombinant MAdCAM-1 and the decrease of this effect by vedolizumab: these parameters have been suggested as predictors of clinical response at 15 weeks in a study on 21 UC patients [67]. Another small study explored serum markers of response in anti-TNF refractory patients, before starting vedolizumab. They found higher levels of IL6 in IBD patients who were subsequently non-responder, of sCD40L in CD non-responder patients and higher osteocalcin levels in UC responders [68]. More recently serum IL6 and IL8 measured at baseline and at week 10 of vedolizumab treatment were suggested as early markers of clinical response at 12 months [69]. The prognostic value of serum biomarkers measured at baseline and at early intervals during vedolizumab therapy was also explored in two studies. In UC, an increase of s-α4β7 an a decrease of s-MAdCAM-1, s-VCAM-1, s-ICAM-1, and s-TNF were found in clinical and endoscopic remitters at 26 weeks [70]. In CD patients, higher early serum levels of s-ICAM-1 and s-VCAM-1 and lower values of s-α4β7 were found in endoscopic remitters [71].
Limited data of transcriptomic and immunologic predictive biomarkers of response to IL23 inhibition are available. IL22 exerts a role of positive regulation on IL23 signalling. In the phase 2a trial of brazikumab (anti-IL 23) in CD patients, higher concentration of IL22 at baseline had an association with increased likelihood of response, even though the association did not test statistically significant [72].
Results from transcriptomic and immunologic studies are resumed in Table 2.
Table 2. Immunological markers.
|
Study |
Immunological Markers |
Outcomes |
|---|---|---|
|
Gaujoux et al. 2019 [58] |
Higher expression of TREM-1 and CCR2-CCL7 in intestinal biopsies |
Nonresponse to anti-TNF treatment |
|
Verstockt et al. 2019 [53] |
Lower expression of TREM-1 in whole blood and intestinal biopsies, lower concentration in serum |
Mucosal healing in patients treated with anti-TNF |
|
Atreya et al. 2014 [60] |
Higher number of mTNF+ cells in intestinal biopsies |
Short term (12 weeks) response to adalimumab |
|
Jessen et al. 2020 [61] |
TNF production > 500 pg/mL by PBMC stimulated with LPS |
Clinical response to infliximab at week 6 |
|
West et al. 2017 [62] |
Higher expression of OSM in intestinal biopsies |
Refractoriness to infliximab (at weeks 8 and 30) and golimumab (at week 6) |
|
Bertani et al. 2020 [63] |
Lower serum concentration of OSM |
Mucosal healing at week 54 in infliximab-treated patients |
|
Boden et al. 2018 [65] |
Higher expression of α4β7 on T effector memory cells and NK cells |
Response to vedolizumab |
|
Rath et al. 2017 [66] |
Presence of α4β7+ cells in intestinal mucosa |
Response to anti-integrin therapy |
|
Allner et al. 2020 [67] |
Higher dynamic adhesion of peripheral blood CD4+ T cells to MAdCAM-1 and more pronounced reduction of adhesion following treatment |
Clinical response to vedolizumab in UC patients |
|
Soendergaard et al. 2020 [68] |
Higher serum IL6 Higher serum CD40L Higher serum osteocalcin |
Nonresponse to vedolizumab in IBD patients Nonresponse to vedolizumab in CD patients Response to vedolizumab in UC patients |
|
Bertani et al. 2020 [69] |
Higher serum IL6 and IL8, more pronounced decrease of IL6 after 10 weeks |
Clinical response to vedolizumab after 12 months |
|
Battat et al. 2019 [70] |
Increase of serum α4β7 and decrease of serum MAdCAM-1, VCAM-1, ICAM-1 and TNF |
Clinical and endoscopic remission ate week 26 in vedolizumab-treated patients |
|
Holmer et al. 2020 [71] |
Higher serum VCAM-1 and ICAM-1 and lower serum α4β7 |
Endoscopic remission in vedolizumab-treated patients |
|
Sands et al. 2017 [72] |
Higher serum IL22 |
Clinical response to brazikumab |
TREM-1: triggering receptor expressed on myeloid cells 1; CCR2-CCL7: chemokine receptor type 2–chemokine ligand 7; TNF: tumor necrosis factor; mTNF: membrane tumor necrosis factor; PBMC: peripheral blood mononuclear cells; LPS: lipopolysaccharide; OSM: oncostatin M; NK cells: natural killer cells; IL: interleukin; CD40L: ligand of cluster of differentiation 40; IBD: inflammatory bowel disease; CD: Crohn’s disease; UC: ulcerative colitis; MAdCAM-1: mucosal vascular addressin cell adhesion molecule 1; VCAM-1: vascular cell adhesion molecule 1; ICAM-1: intercellular adhesion molecule 1.
This entry is adapted from the peer-reviewed paper 10.3390/jcm10040853
References
- Kaplan, G.G. The global burden of IBD: From 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 720–727.
- Park, K.T.; Ehrlich, O.G.; Allen, J.I.; Meadows, P.; Szigethy, E.M.; Henrichsen, K.; Kim, S.C.; Lawton, R.C.; Murphy, S.M.; Regueiro, M.; et al. The Cost of Inflammatory Bowel Disease: An Initiative from the Crohn’s & Colitis Foundation. Inflamm. Bowel Dis. 2020, 26, 1–10.
- Harbord, M.; Eliakim, R.; Bettenworth, D.; Karmiris, K.; Katsanos, K.; Kopylov, U.; Kucharzik, T.; Molnár, T.; Raine, T.; Sebastian, S.; et al. Third European Evidence-based Consensus on Diagnosis and Management of Ulcerative Colitis. Part 2: Current Management. J. Crohn’s Colitis 2017, 11, 769–784.
- Torres, J.; Bonovas, S.; Doherty, G.; Kucharzik, T.; Gisbert, J.P.; Raine, T.; Adamina, M.; Armuzzi, A.; Bachmann, O.; Bager, P.; et al. ECCO Guidelines on Therapeutics in Crohn’s Disease: Medical Treatment. J. Crohn’s Colitis 2020, 14, 4–22.
- Sandborn, W.J.; Su, C.; Sands, B.E.; D’Haens, G.R.; Vermeire, S.; Schreiber, S.; Danese, S.; Feagan, B.G.; Reinisch, W.; Niezychowski, W.; et al. Tofacitinib as Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2017, 376, 1723–1736.
- Verdier, J.; Bègue, B.; Cerf-Bensussan, N.; Ruemmele, F.M. Compartmentalized Expression of Th1 and Th17 Cytokines in Pediatric Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2012, 18, 1260–1266.
- Atreya, R.; Neurath, M.F. Mechanisms of molecular resistance and predictors of response to biological therapy in inflammatory bowel disease. Lancet Gastroenterol. Hepatol. 2018, 3, 790–802.
- Zorzi, F.; Monteleone, I.; Sarra, M.; Calabrese, E.; Marafini, I.; Cretella, M.; Sedda, S.; Biancone, L.; Pallone, F.; Monteleone, G. Distinct Profiles of Effector Cytokines Mark the Different Phases of Crohn’s Disease. PLoS ONE 2013, 8, e54562.
- Singh, S.; Murad, M.H.; Fumery, M.; Dulai, P.S.; Sandborn, W.J. First- and Second-Line Pharmacotherapies for Patients with Moderate to Severely Active Ulcerative Colitis: An Updated Network Meta-Analysis. Clin. Gastroenterol. Hepatol. 2020, 18, 2179–2191.e6.
- Singh, S.; Fumery, M.; Sandborn, W.J.; Murad, M.H. Systematic review and network meta-analysis: First- and second-line biologic therapies for moderate-severe Crohn’s disease. Aliment. Pharmacol. Ther. 2018, 48, 394–409.
- Singh, S.; George, J.; Boland, B.S.; Casteele, N.V.; Sandborn, W.J. Primary Non-Response to Tumor Necrosis Factor Antagonists is Associated with Inferior Response to Second-line Biologics in Patients with Inflammatory Bowel Diseases: A Systematic Review and Meta-analysis. J. Crohn’s Colitis 2018, 12, 635–643.
- Ben-Horin, S.; Kopylov, U.; Chowers, Y. Optimizing anti-TNF treatments in inflammatory bowel disease. Autoimmun. Rev. 2014, 13, 24–30.
- Gisbert, J.P.; Chaparro, M. Predictors of Primary Response to Biologic Treatment [Anti-TNF, Vedolizumab, and Ustekinumab] in Patients with Inflammatory Bowel Disease: From Basic Science to Clinical Practice. J. Crohn’s Colitis 2020, 14, 694–709.
- Hamdeh, S.; Aziz, M.; Altayar, O.; Olyaee, M.; Murad, M.H.; Hanauer, S.B. Early vs Late Use of Anti-TNFa Therapy in Adult Patients with Crohn Disease: A Systematic Review and Meta-Analysis. Inflamm. Bowel Dis. 2020, 26, 1808–1818.
- Ungaro, R.C.; Aggarwal, S.; Topaloglu, O.; Lee, W.-J.; Clark, R.; Colombel, J.-F. Systematic review and meta-analysis: Efficacy and safety of early biologic treatment in adult and paediatric patients with Crohn’s disease. Aliment. Pharmacol. Ther. 2020, 51, 831–842.
- Singh, N.; Rabizadeh, S.; Jossen, J.; Pittman, N.; Check, M.; Hashemi, G.; Phan, B.L.; Hyams, J.S.; Dubinsky, M.C. Multi-Center Experience of Vedolizumab Effectiveness in Pediatric Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2016, 22, 2121–2126.
- Vermeire, S.; Louis, E.; Carbonez, A.; Van Assche, G.; Noman, M.; Belaiche, J.; De Vos, M.; Van Gossum, A.; Pescatore, P.; Fiasse, R.; et al. Demographic and clinical parameters influencing the short-term outcome of anti-tumor necrosis factor (infliximab) treatment in Crohn’s disease. Am. J. Gastroenterol. 2002, 97, 2357–2363.
- Sprakes, M.B.; Ford, A.C.; Warren, L.; Greer, D.; Hamlin, J. Efficacy, tolerability, and predictors of response to infliximab therapy for Crohn’s disease: A large single centre experience. J. Crohn’s Colitis 2012, 6, 143–153.
- Zorzi, F.; Zuzzi, S.; Onali, S.; Calabrese, E.; Condino, G.; Petruzziello, C.; Ascolani, M.; Pallone, F.; Biancone, L. Efficacy and safety of infliximab and adalimumab in Crohn’s disease: A single centre study. Aliment. Pharmacol. Ther. 2012, 35, 1397–1407.
- Kiss, L.S.; Szamosi, T.; Molnár, T.; Miheller, P.; Lakatos, L.; Vincze, A.; Palatka, K.; Barta, Z.; Gasztonyi, B.; Salamon, A.; et al. Early clinical remission and normalisation of CRP are the strongest predictors of efficacy, mucosal healing and dose escalation during the first year of adalimumab therapy in Crohn’s disease. Aliment. Pharmacol. Ther. 2011, 34, 911–922.
- Bertani, L.; Tricò, D.; Pugliese, D.; Privitera, G.; Linsalata, G.; Zanzi, F.; Mumolo, M.G.; Barberio, B.; Monzani, F.; Marchi, S.; et al. Serum triiodothyronine-to-thyroxine (T3/T4) ratio predicts therapeutic outcome to biological therapies in elderly IBD patients. Aliment. Pharmacol. Ther. 2020, 53.
- Colombel, J.; Sandborn, W.J.; Rutgeerts, P.; Enns, R.; Hanauer, S.B.; Panaccione, R.; Schreiber, S.; Byczkowski, D.; Li, J.; Kent, J.D.; et al. Adalimumab for Maintenance of Clinical Response and Remission in Patients with Crohn’s Disease: The CHARM Trial. Gastroenterology 2007, 132, 52–65.
- Peters, C.P.; Eshuis, E.J.; Toxopeüs, F.M.; Hellemons, M.E.; Jansen, J.M.; D’Haens, G.R.A.M.; Fockens, P.; Stokkers, P.C.F.; Tuynman, H.A.R.E.; Van Bodegraven, A.A.; et al. Adalimumab for Crohn’s disease: Long-term sustained benefit in a population-based cohort of 438 patients. J. Crohn’s Colitis 2014, 8, 866–875.
- Jürgens, M.; John, J.M.M.; Cleynen, I.; Schnitzler, F.; Fidder, H.; Van Moerkercke, W.; Ballet, V.; Noman, M.; Hoffman, I.; Van Assche, G.; et al. Levels of C-reactive Protein Are Associated with Response to Infliximab Therapy in Patients with Crohn’s Disease. Clin. Gastroenterol. Hepatol. 2011, 9, 421–427.e1.
- Reinisch, W.; Wang, Y.; Oddens, B.J.; Link, R. C-reactive protein, an indicator for maintained response or remission to infliximab in patients with Crohn’s disease: A post-hoc analysis from ACCENT I. Aliment. Pharmacol. Ther. 2012, 35, 568–576.
- Ferrante, M.; Vermeire, S.; Fidder, H.; Schnitzler, F.; Noman, M.; Van Assche, G.; De Hertogh, G.; Hoffman, I.; D’Hoore, A.; Van Steen, K.; et al. Long-term outcome after infliximab for refractory ulcerative colitis. J. Crohn’s Colitis 2008, 2, 219–225.
- Arias, M.T.; Casteele, N.V.; Vermeire, S.; Overstraeten, A.D.B.V.; Billiet, T.; Baert, F.; Wolthuis, A.; Van Assche, G.; Noman, M.; Hoffman, I.; et al. A Panel to Predict Long-term Outcome of Infliximab Therapy for Patients with Ulcerative Colitis. Clin. Gastroenterol. Hepatol. 2015, 13, 531–538.
- Reinisch, W.; Sandborn, W.J.; Hommes, D.W.; D’Haens, G.; Hanauer, S.; Schreiber, S.; Panaccione, R.; Fedorak, R.N.; Tighe, M.B.; Huang, B.; et al. Adalimumab for induction of clinical remission in moderately to severely active ulcerative colitis: Results of a randomised controlled trial. Gut 2011, 60, 780–787.
- Guidi, L.; Marzo, M.; Andrisani, G.; Felice, C.; Pugliese, D.; Mocci, G.; Nardone, O.; De Vitis, I.; Papa, A.; Rapaccini, G.; et al. Faecal calprotectin assay after induction with anti-Tumour Necrosis Factor α agents in inflammatory bowel disease: Prediction of clinical response and mucosal healing at one year. Dig. Liver Dis. 2014, 46, 974–979.
- Gisbert, J.P.; Marín, A.C.; McNicholl, A.G.; Chaparro, M. Systematic review with meta-analysis: The efficacy of a second anti-TNF in patients with inflammatory bowel disease whose previous anti-TNF treatment has failed. Aliment. Pharmacol. Ther. 2015, 41, 613–623.
- Casanova, M.J.; Chaparro, M.; Mínguez, M.; Ricart, E.; Taxonera, C.; García-López, S.; Guardiola, J.; Román, A.L.-S.; Iglesias, E.; Beltrán, B.; et al. Effectiveness and Safety of the Sequential Use of a Second and Third Anti-TNF Agent in Patients with Inflammatory Bowel Disease: Results from the Eneida Registry. Inflamm. Bowel Dis. 2020, 26, 606–616.
- Papamichael, K.; Rivals-Lerebours, O.; Billiet, T.; Casteele, N.V.; Gils, A.; Ferrante, M.; Van Assche, G.; Rutgeerts, P.; Mantzaris, G.J.; Peyrin-Biroulet, L.; et al. Long-Term Outcome of Patients with Ulcerative Colitis and Primary Non-response to Infliximab. J. Crohn’s Colitis 2016, 10, 1015–1023.
- Park, S.C.; Jeen, Y.T. Genetic Studies of Inflammatory Bowel Disease-Focusing on Asian Patients. Cells 2019, 8, 404.
- Bek, S.; Nielsen, J.V.; Bojesen, A.B.; Franke, A.; Bank, S.; Vogel, U.; Andersen, V. Systematic review: Genetic biomarkers associated with anti-TNF treatment response in inflammatory bowel diseases. Aliment. Pharmacol. Ther. 2016, 44, 554–567.
- Tong, Q.; Zhao, L.; Qian, X.-D.; Zhang, L.-L.; Xu, X.; Dai, S.-M.; Cai, Q.; Zhao, D. Association ofTNF-α polymorphism with prediction of response to TNF blockers in spondyloarthritis and inflammatory bowel disease: A meta-analysis. Pharmacogenomics 2013, 14, 1691–1700.
- Bank, S.; Andersen, P.S.; Burisch, J.; Pedersen, N.; Roug, S.; Galsgaard, J.; Turino, S.Y.; Brodersen, J.B.; Rashid, S.; Rasmussen, B.K.; et al. Associations between functional polymorphisms in the NFκB signaling pathway and response to anti-TNF treatment in Danish patients with inflammatory bowel disease. Pharm. J. 2014, 14, 526–534.
- Jürgens, M.; Laubender, R.P.; Hartl, F.; Weidinger, M.; Seiderer, J.; Wagner, J.; Wetzke, M.; Beigel, F.; Pfennig, S.; Stallhofer, J.; et al. Disease Activity, ANCA, and IL23R Genotype Status Determine Early Response to Infliximab in Patients with Ulcerative Colitis. Am. J. Gastroenterol. 2010, 105, 1811–1819.
- Sazonovs, A.; Kennedy, N.A.; Moutsianas, L.; Heap, G.A.; Rice, D.L.; Reppell, M.; Bewshea, C.M.; Chanchlani, N.; Walker, G.J.; Perry, M.H.; et al. HLA-DQA1*05 Carriage Associated with Development of Anti-Drug Antibodies to Infliximab and Adalimumab in Patients with Crohn’s Disease. Gastroenterology 2020, 158, 189–199.
- Billiet, T.; Casteele, N.V.; Van Stappen, T.; Princen, F.; Singh, S.; Gils, A.; Ferrante, M.; Van Assche, G.; Cleynen, I.; Vermeire, S. Immunogenicity to infliximab is associated with HLA-DRB1. Gut 2015, 64, 1344–1345.
- Louis, E.; El Ghoul, Z.; Vermeire, S.; Dall’Ozzo, S.; Rutgeerts, P.; Paintaud, G.; Belaiche, J.; De Vos, M.; Van Gossum, A.; Colombel, J.F.; et al. Association between polymorphism in IgG Fc receptor IIIa coding gene and biological response to infliximab in Crohn’s disease. Aliment. Pharmacol. Ther. 2004, 19, 511–519.
- Niess, J.H.; Klaus, J.; Stephani, J.; Pflüger, C.; Degenkolb, N.; Spaniol, U.; Mayer, B.; Lahr, G.; Von Boyen, G.B.T. NOD2 Polymorphism Predicts Response to Treatment in Crohn’s Disease—First Steps to a Personalized Therapy. Dig. Dis. Sci. 2012, 57, 879–886.
- Juanola, O.; Moratalla, A.; Gutierrez, A.; Sempere, L.; Zapater, P.; Giménez, P.; Almenta, I.; Peiró, G.; González-Navajas, J.M.; Such, J.F.; et al. Anti-TNF-alpha loss of response is associated with a decreased percentage of FoxP3+ T cells and a variant NOD2 genotype in patients with Crohn’s disease. J. Gastroenterol. 2015, 50, 758–768.
- Schäffler, H.; Geiss, D.; Gittel, N.; Rohde, S.; Huth, A.; Glass, Ä; Brandhorst, G.; Jaster, R.; Lamprecht, G. Mutations in theNOD2gene are associated with a specific phenotype and lower anti-tumor necrosis factor trough levels in Crohn’s disease. J. Dig. Dis. 2018, 19, 678–684.
- Koder, S.; Repnik, K.; Ferkolj, I.; Pernat, C.; Skok, P.; Weersma, R.; Potočnik, U. Genetic polymorphism inATG16L1gene influences the response to adalimumab in Crohn’s disease patients. Pharmacogenomics 2015, 16, 191–204.
- Hlavaty, T.; Ferrante, M.; Henckaerts, L.; Pierik, M.; Rutgeerts, P.; Vermeire, S. Predictive Model for the Outcome of Infliximab Therapy in Crohn’s Disease Based on Apoptotic Pharmacogenetic Index and Clinical Predictors. Inflamm. Bowel Dis. 2007, 13, 372–379.
- Barber, G.E.; Yajnik, V.; Khalili, H.; Giallourakis, C.; Garber, J.; Xavier, R.; Ananthakrishnan, A.N. Genetic Markers Predict Primary Non-Response and Durable Response to Anti-TNF Biologic Therapies in Crohn’s Disease. Am. J. Gastroenterol. 2016, 111, 1816–1822.
- Burke, K.E.; Khalili, H.; Garber, J.; Haritunians, T.; McGovern, D.P.; Xavier, R.J.; Ananthakrishnan, A.N. Genetic Markers Predict Primary Nonresponse and Durable Response to Anti–Tumor Necrosis Factor Therapy in Ulcerative Colitis. Inflamm. Bowel Dis. 2018, 24, 1840–1848.
- Wang, M.-H.; Friton, J.J.; Raffals, L.E.; Leighton, J.A.; Pasha, S.F.; Picco, M.F.; Cushing, K.C.; Monroe, K.; Nix, B.D.; Newberry, R.D.; et al. Novel Genetic Risk Variants Can Predict Anti-TNF Agent Response in Patients with Inflammatory Bowel Disease. J. Crohn’s Colitis 2019, 13, 1036–1043.
- Atreya, I.; Atreya, R.; Neurath, M.F. NF-κB in inflammatory bowel disease. J. Intern. Med. 2008, 263, 591–596.
- Vermeire, S.; Gils, A.; Accossato, P.; Lula, S.; Marren, A. Immunogenicity of biologics in inflammatory bowel disease. Ther. Adv. Gastroenterol. 2018, 11.
- Colombel, J.-F.; Adedokun, O.J.; Gasink, C.; Gao, L.-L.; Cornillie, F.J.; D’Haens, G.R.; Rutgeerts, P.J.; Reinisch, W.; Sandborn, W.J.; Hanauer, S.B. Combination Therapy with Infliximab and Azathioprine Improves Infliximab Pharmacokinetic Features and Efficacy: A Post Hoc Analysis. Clin. Gastroenterol. Hepatol. 2019, 17, 1525–1532.e1.
- Arijs, I.; Li, K.; Toedter, G.; Quintens, R.; Van Lommel, L.; Van Steen, K.; Leemans, P.; De Hertogh, G.; Lemaire, K.; Ferrante, M.; et al. Mucosal gene signatures to predict response to infliximab in patients with ulcerative colitis. Gut 2009, 58, 1612–1619.
- Verstockt, B.; Verstockt, S.; Creyns, B.; Tops, S.; Van Assche, G.; Gils, A.; Ceuppens, J.L.; Vermeire, S.; Ferrante, M.; Breynaert, C. Mucosal IL13RA2 expression predicts nonresponse to anti-TNF therapy in Crohn’s disease. Aliment. Pharmacol. Ther. 2019, 49, 572–581.
- Ogura, Y.; Bonen, D.K.; Inohara, N.; Nicolae, D.L.; Chen, F.F.; Ramos, R.; Britton, H.; Moran, T.; Karaliuskas, R.; Duerr, R.H.; et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nat. Cell Biol. 2001, 411, 603–606.
- Weersma, R.K.; Stokkers, P.C.F.; Van Bodegraven, A.A.; Van Hogezand, R.A.; Verspaget, H.W.; De Jong, D.J.; Van Der Woude, C.J.; Oldenburg, B.; Linskens, R.K.; Festen, E.A.M.; et al. Molecular prediction of disease risk and severity in a large Dutch Crohn’s disease cohort. Gut 2009, 58, 388–395.
- Vermeire, S.; O’Byrne, S.; Keir, M.; Williams, M.; Lu, T.T.; Mansfield, J.C.; Lamb, C.A.; Feagan, B.G.; Panes, J.; Salas, A.; et al. Etrolizumab as induction therapy for ulcerative colitis: A randomised, controlled, phase 2 trial. Lancet 2014, 384, 309–318.
- Tew, G.W.; Hackney, J.A.; Gibbons, D.; Lamb, C.A.; Luca, D.; Egen, J.G.; Diehl, L.; Anderson, J.E.; Vermeire, S.; Mansfield, J.C.; et al. Association Between Response to Etrolizumab and Expression of Integrin αE and Granzyme A in Colon Biopsies of Patients with Ulcerative Colitis. Gastroenterology 2016, 150, 477–487.e9.
- Gaujoux, R.; Starosvetsky, E.; Maimon, N.; Vallania, F.; Bar-Yoseph, H.; Pressman, S.; Weisshof, R.; Goren, I.; Rabinowitz, K.; Waterman, M.; et al. Cell-centred meta-analysis reveals baseline predictors of anti-TNFα non-response in biopsy and blood of patients with IBD. Gut 2019, 68, 604–614.
- Verstockt, B.; Verstockt, S.; Dehairs, J.; Ballet, V.; Blevi, H.; Wollants, W.-J.; Breynaert, C.; Van Assche, G.; Vermeire, S.; Ferrante, M. Low TREM1 expression in whole blood predicts anti-TNF response in inflammatory bowel disease. EBioMedicine 2019, 40, 733–742.
- Atreya, R.; Neumann, H.; Neufert, C.; Waldner, M.J.; Billmeier, U.; Zopf, Y.; Willma, M.; App, C.; Münster, T.; Kessler, H.; et al. In vivo imaging using fluorescent antibodies to tumor necrosis factor predicts therapeutic response in Crohn’s disease. Nat. Med. 2014, 20, 313–318.
- Jessen, B.; Rodriguez-Sillke, Y.; Sonnenberg, E.; Schumann, M.; Kruglov, A.; Freise, I.; Schmidt, F.; Maul, J.; Kühl, A.A.; Glauben, R.; et al. Level of Tumor Necrosis Factor Production by Stimulated Blood Mononuclear Cells Can Be Used to Predict Response of Patients with Inflammatory Bowel Diseases to Infliximab. Clin. Gastroenterol. Hepatol. 2020.
- West, N.R.; Hegazy, A.N.; Owens, B.M.J.; Bullers, S.J.; Linggi, B.; Buonocore, S.; Coccia, M.; Görtz, D.; This, S.; Stockenhuber, K.; et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor–neutralizing therapy in patients with inflammatory bowel disease. Nat. Med. 2017, 23, 579–589.
- Bertani, L.; Fornai, M.; Fornili, M.; Antonioli, L.; Benvenuti, L.; Tapete, G.; Svizzero, G.B.; Ceccarelli, L.; Mumolo, M.G.; Baglietto, L.; et al. Serum oncostatin M at baseline predicts mucosal healing in Crohn’s disease patients treated with infliximab. Aliment. Pharmacol. Ther. 2020, 52, 284–291.
- Ungar, B.; Kopylov, U.; Yavzori, M.; Fudim, E.; Picard, O.; Lahat, A.; Coscas, D.; Waterman, M.; Haj-Natour, O.; Orbach-Zingboim, N.; et al. Association of Vedolizumab Level, Anti-Drug Antibodies, and α4β7 Occupancy with Response in Patients with Inflammatory Bowel Diseases. Clin. Gastroenterol. Hepatol. 2018, 16, 697–705.e7.
- Boden, E.K.; Shows, D.M.; Chiorean, M.V.; Lord, J.D. Identification of Candidate Biomarkers Associated with Response to Vedolizumab in Inflammatory Bowel Disease. Dig. Dis. Sci. 2018, 63, 2419–2429.
- Rath, T.; Bojarski, C.; Neurath, M.; Atreya, R. Molecular imaging of mucosal α4β7 integrin expression with the fluorescent anti-adhesion antibody vedolizumab in Crohn’s disease. Gastrointest. Endosc. 2017, 86, 406–408.
- Allner, C.; Melde, M.; Becker, E.; Fuchs, F.; Mühl, L.; Klenske, E.; Müller, L.; Morgenstern, N.; Fietkau, K.; Hirschmann, S.; et al. Baseline levels of dynamic CD4+ T cell adhesion to MAdCAM-1 correlate with clinical response to vedolizumab treatment in ulcerative colitis: A cohort study. BMC Gastroenterol. 2020, 20, 1–9.
- Soendergaard, C.; Seidelin, J.B.; Steenholdt, C.; Nielsen, O.H. Putative biomarkers of vedolizumab resistance and underlying inflammatory pathways involved in IBD. BMJ Open Gastroenterol. 2018, 5, e000208.
- Bertani, L.; Caviglia, G.P.; Antonioli, L.; Pellicano, R.; Sharmila, F.; Astegiano, M.; Saracco, G.M.; Bugianesi, E.; Blandizzi, C.; Costa, F.; et al. Serum Interleukin-6 and -8 as Predictors of Response to Vedolizumab in Inflammatory Bowel Diseases. J. Clin. Med. 2020, 9, 1323.
- Battat, R.; Dulai, P.S.; Casteele, N.V.; Evans, E.; Hester, K.D.; Webster, E.; Jain, A.; Proudfoot, J.A.; Mairalles, A.; Neill, J.; et al. Biomarkers Are Associated with Clinical and Endoscopic Outcomes with Vedolizumab Treatment in Ulcerative Colitis. Inflamm. Bowel Dis. 2019, 25, 410–420.
- Holmer, A.K.; Battat, R.; Dulai, P.S.; Casteele, N.V.; Nguyen, N.; Jain, A.; Miralles, A.; Neill, J.; Le, H.; Singh, S.; et al. Biomarkers are associated with clinical and endoscopic outcomes with vedolizumab treatment in Crohn’s disease. Ther. Adv. Gastroenterol. 2020, 13.
- Sands, B.E.; Chen, J.; Feagan, B.G.; Penney, M.; Rees, W.A.; Danese, S.; Higgins, P.D.; Newbold, P.; Faggioni, R.; Patra, K.; et al. Efficacy and Safety of MEDI2070, an Antibody Against Interleukin 23, in Patients with Moderate to Severe Crohn’s Disease: A Phase 2a Study. Gastroenterology 2017, 153, 77–86.e6.