Myeloid malignancies present with a distinct metabolomic signature. Targeting metabolic pathways has become a potent therapeutic strategy for this group of disorders. The biological basis of this approach resides in the metabolic regulation of normal hematopoiesis and their alterations.
- myeloid malignancies
- TET2 mutations
- IDH1/2 mutations
- venetoclax
1. Introduction
During normal cellular development, metabolism is one of the principal modalities to produce energy. In fact, from cell birth to differentiation, cell metabolism, together with a switch in transcriptional factors and cytokine release in some instances, dictate cell fate. As an example, hematopoietic stem cells (HSCs) adapt the balance between survival and quiescence by keeping a low oxygen status, stabilizing the hypoxia-inducible factor (HIF-1α), and stimulate glycolysis activating enzymes regulating glucose uptake (glucose transporter 1, Glut1) and pyruvate synthesis (Lactate Dehydrogenase A, LDHA). The main demand of HSCs is maintaining themselves in a quiescent status which is easily reached by using low energetic power through glycolysis [1]. Studies have shown how changes in metabolism, specifically in the glycolytic process, are notably present in acute myeloid leukemia (AML) [2][3][4]. Indeed history goes back to the first measurements of tumor metabolism when Nobel Prize in Physiology and Medicine Otto Warburg discovered that malignant cells favor the production of lactic acid (anaerobic glycolysis) in the TCA cycle as a source of ATP production. Leukemia-initiating cells (LICs) have an increased glycolytic flux mediated by AMP-activated kinase (AMPK) activation and decreased levels of autophagic activity. Often, increased glycolytic activity has been associated with resistance to pharmacologic agents [5]. Moreover, the high flux of glucose has been linked to increased glucose-6-phosphate dehydrogenase (G6PD) and consequently to unfavorable prognosis. Indeed FLT3 inhibitors have been shown to inactivate G6PD in FLT3 mutant AML [6]. Because of the high demand for glucose, frequently AML cells switch to the use of fructose as an energy source, suggesting that the inhibition of fructose uptake might be a way to starve AML cells and reduce their malignant potential.
Leukemia stem cells (LSC) rely mainly on oxidative phosphorylation (OXPHOS) through BCL-2, suggesting the basis to target BCL-2-dependent pathways [7]. Another modality of controlling LSCs to use OXPHOS is through glutamine metabolism. In fact, inhibition of the conversion of glutamine in glutamate by blocking glutaminases is able to arrest leukemic activity, and targeting glutamine conversion has been found efficient in synergy with BCL-2 inhibition [8]. Overproduction of endogenous reactive oxygen species is also another option to help AML cells to promote blast proliferation. Studies aiming to describe the global metabolic profiles and redox status of AML cells have shown differences in metabolic spectra between diverse cytogenetic and molecular subtypes [9][10]. Moreover, the presence of specific metabolites has been associated with prognosis and identified as markers of aggressiveness (phosphocholine and phosphoethanolamine) and chemoresistance (overexpression of glutathione). Altogether the deregulation of metabolic pathways in AML might contribute to therapy-resistance and relapse.
Targeting the metabolic pathway is feasible but mostly depends on a fine and specific targeting of only altered processes, given the importance of metabolic functions in normal cells. In this line, the concomitant use of metabolic pathways inhibitors and differentiation agents (BCL-2 inhibitors and hypomethylating agents, HMA) represents a promising avenue in drug discovery. Recently the combination of the data obtained from the Reactome and KEGG pathway and The Cancer Genome Atlas has defined the interaction between metabolic pathways and molecular and transcriptomic signatures in all cancers, including myeloid malignancies.
Here, author provide examples of the link between altered metabolism and leukemogenic potential, focusing on some of the major mutations in myeloid genes (e.g., TET2 and IDH1/2) generating important biological consequences in a variety of cellular processes. Moreover, author will also review available therapeutic options and examples of dysregulation in genes involved in metabolism (Figure 1 and Figure 2).
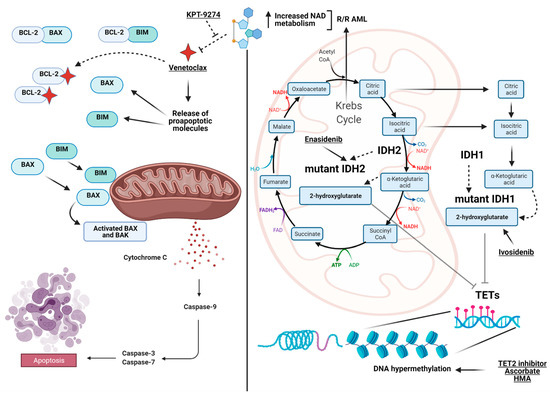
Figure 1. View at a glance of the landscape of the metabolic interactions between genes and pathways discussed in the manuscript. On the left, the intrinsic apoptotic pathway highlighted by BCL-2 as the pivotal player (targeted by Venetoclax) and the interaction with BAX and BIM to initiate the caspases cascade to trigger apoptosis. On the right, the interaction between the Krebs cycle (TCA), the convergence of IDH1/2 generating α-ketoglutarate, 2-hydroxyglutarate and the TET family of genes, which finally impacts DNA methylation. Included are also the available drugs used to target the depicted pathways. On the top, in relapsed/refractory (R/R) acute myeloid leukemia (AML), the interconnection between the increase in NAD metabolism generated by alteration of oxidative phosphorylation and Venetoclax resistance is shown together with proposed actionable target agents (e.g., KPT-9274) inhibiting Nicotinamide Phosphoribosyltransferase (NAMPT). Images were generated using BioRender.
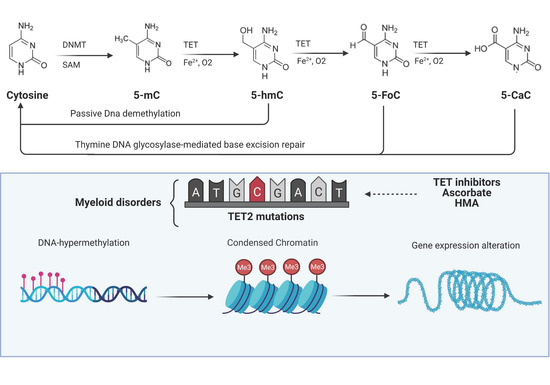
Figure 2. TET functions and their alterations in myeloid malignancies. DNA methyltransferases (DNMTs) initiate cytosine methylation with conversion to 5-methylcytosine (5-mc). TET proteins progressively oxidize 5-mC to 5-hydroxymethylcytosine (5-hmC), 5-formylcytosine (5-FoC), and 5-carboxylcytosine (5-CaC) creating a pool of TET-oxidized products (TDOP). 5-hmC can be reverted to cytosine via passive dilution while 5-FoC and 5-CaC via thymine DNA glycosylase-mediated base excision repair. Somatic TET2 mutations create an imbalance in cellular DNA methylation through the disruption of the aforementioned mechanism with alteration of chromatin and thereby consequences on expression of genes regulating cell division and self-renewal. Images were generated using BioRender.
2. The Metabolomics of TET Family of Genes
The Ten-Eleven Translocation (TET) gene family (TET1, TET2, and TET3) encodes for dioxygenase enzymes catalyzing the conversion of 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC) [11]. The first discovered gene was TET1, identified as a partner of mixed-lineage leukemia (MLL/KMT2A) gene in a case of infant AML carrying a t(10:11) (q22;q23) translocation [12][13]. The finding of the dioxygenase function of TET1 led to the identification of the other two members of the TET family, TET2 and TET3 [14][15].
The carboxyl (C)-terminus of all three TET family members harbors the oxygenase catalytic domain and the binding sites for 2-oxoglutarate (α-KG) and Fe2+, which are both critical cofactors for TET oxidative activity [16]. TET proteins progressively oxidize 5-mC to 5-hmC, 5-formylcytosine (5-fC), and 5-carboxylcytosine (5-caC), creating a pool of TET-dependent 5-mC-DNA oxidation products (TDOP) which ultimately can be reverted to cytosine via thymine DNA glycosylase-mediated base excision repair [16][17].(Figure 2)
Therefore, TET proteins are critical regulators of DNA demethylation via the generation of 5-hmC, ultimately leading to enhanced gene expression and transcription profiles responsible for cell proliferation and survival [18][19][20]. As the role of the TET family of genes is highly crucial during cell development, any impairment either by genetic lesions or altered expression might impact the normal fate of the cells. Indeed, high levels of expression of TET family proteins and, as a consequence, of 5-hmC have also been found to be crucial for embryonic stem cells, determining cell fate and development with redundant functions [21][22]. As demonstrated by knockout (KO) experiments in mice, triple Tet1/2/3 inactivation led to embryonic lethality while other Tet1 and/or Tet2 KO configurations were not completely disruptive [21][23][24]. TET2 haploinsufficiency altered HSC reprogramming into induced pluripotent stem cells, as demonstrated by an experiment of TET2 silencing via short hairpin RNA with different consequences according to the type of TET2 mutation present [25]. Thus, while the pivotal role of TET2 mutations is underlined by their high frequency in myeloid disorders, including AML, the lack of molecular lesions in the homologous TET1/TET3 genes is probably explicable with a distinct tissue-specific expression and differences in the specific metabolic consequences derived from their imbalance [26].
2.1. TET2: A Pivotal Gene in Myeloid Malignancies
Somatic TET2 mutations are commonly found in myeloid malignancies (MN) at frequencies varying according to disease subtypes. In myelodysplastic syndromes (MDS) and AML, TET2 mutations are present in 20%–30% of cases, ranging up to 50% in patients affected by chronic myelomonocytic leukemia (CMML) [27][28][29]. Mutations are mainly loss-of-function (either frameshift or nonsense) affecting the coding region or missense mutations mapping in critical sites required for the enzymatic activity [30]. The prognostic role of TET2 lesions has been controversial because of their high frequency, their heterogeneity, and the variability of the concurrent genetic lesions, all characteristics contributing to shaping the fate of individual patients and ultimately precluding a clear genotype/phenotype association [30]. Moreover, the discovery of mutations in myeloid genes in normal individual (referred to as CHIP, clonal hematopoiesis of indeterminate potential) at frequencies linearly correlated with age (age-related clonal hematopoiesis, ARCH) shed light on the process of myeloid evolution and provided clues on clonality and subclonal hierarchies in myeloid malignancies [31]. The occurrence of TET2 mutations in CHIP/ARCH is another confirmation of the importance of this gene in cell development. Together with additional sex combs-like 1 (ASXL1) and DNA methyltransferase 3 alpha (DNMT3A) genes, TET2 represents one of the most frequently mutated genes in CHIP/ARCH so that the three genes are referred to with the acronym DAT (DNMT3A/ASXL1/TET2) [31]. This finding, as well as the ubiquitous presence of TET2 mutations in hematological malignancies, indicate that TET2 lesions are mainly ancestral events occurring early in the course of the disease and contribute to the creation of a so-called “mutator phenotype”, by giving to the clone a proclivity for the acquisition of additional molecular lesions [32]. Indeed, our group demonstrated that in a cohort of 4930 patients with MNs, 1205 (24%) carried TET2 mutations, which were ancestral and probably deriving from TET2-mutant CHIP in >40% of cases [30]. Subclonal acquisition of new leukemogenic events was identified as a facilitating condition for later myeloid progression and disease phenotypic determination (dysplastic vs. proliferative), as underlined by the higher number of secondary mutational events in TET2-mutant vs. TET2 wild-type MN and in Tet2 murine models [30]. Of note, progressive TET2 inactivation was associated with disease progression and poor survival outcomes as demonstrated by the aforementioned tendency of TET2 mutant cases to accumulate additional hits in the same gene either resulting in biallelic mutations, deletions in hemizygous configurations, or uniparental disomies (UPDs) with homozygous mutations, with the latter two groups registering a negative impact on survival [30]. As a matter of fact, in another study focusing only on biallelic TET2-mutant MN, we confirmed that biallelic inactivation is frequently observed in MN and that this configuration is a typical feature of older patients with monocytosis (also outside the context of an overt CMML diagnosis), CMML, normal karyotype, and lower-risk disease. Therefore, biallelic TET2 inactivation led to a disease phenotype skewed towards higher odds of monocytic vs. dysplastic features consistent with its prevalence in CMML [33]. Development of a CMML-like disease in mice has been reported in the literature, confirming the role of TET2 in driving differentiation pressure towards a myelo-monocytic lineage consistent with the high frequency of TET2 mutations in patients with CMML [34][35][36]. The association of TET2 biallelic cases with lower-risk disease, rather than with a more aggressive phenotype as in the case of other TET2 double-hits configurations (deletions in hemizygous configurations or UPDs with homozygous mutations), may be explainable with compensatory TET1/TET3 stabilizing functions or acetylation mechanisms leading to increased TET2 protein stability [33]. The p300-mediated acetylation of key lysine residues (K110 and K111) at the amino (N)-terminus of TET2 enhances its functions and protects against DNA methylation during oxidative stress interacting with DNA methyltransferases (DNMTs) and regulating 5-mc/5-hmC balance [37]. The importance of metabolic events following TET2 loss-of-function mutations in the pathogenesis of MN is also outlined by data showing its general down-regulation in patients with MN and the down-regulation of its family members [38]. Of note, low expression levels of the TET2 gene may be found independently of the presence of its mutations [39][40][41]. Indeed TET2 expression and 5-hmC levels were found also decreased in pediatric MDS cases, a population known to be rarely mutated in TET2 gene [41]. TET2 expression levels have also been identified as a predictive and a prognostic biomarker in cytogenetically normal (CN)-AML [42]. Indeed, low TET2 expression had a negative impact on overall survival (OS) in both non-M3 and CN-AML (p = 0.016 and 0.044, respectively), although multivariable analysis confirmed these results only for the CN group [42]. Conversely, higher expression of TET1 at diagnosis was associated with poor clinical outcomes in a cohort of 360 CN-AML patients [43]. Finally, a recent study demonstrated that high TET3 expression was an independent factor for better OS and disease-free survival (DFS) in AML [44]. Of note, patients with lower TET3 expression undergoing hematopoietic stem cell transplant (HSCT) showed better OS and DFS than those who did not proceed to HSCT [44].
2.2. TET2 as an Actionable Target
Altogether, these data provide evidence that the TET family of genes, and in particular TET2, are actionable therapeutic targets in MN. In this line, recent findings suggested that ascorbic acid (AA) was able to restore some TET2 metabolic activities in vitro [45][46][47]. In addition, AA depletion in mice cooperated with Flt3-ITD mutations to accelerate leukemogenesis, whereas the reintroduction of dietary AA reversed this phenomenon by promoting Tet functions [46]. Moreover, AA was able to restore 5-hmC formation, drive DNA hypomethylation and expression of a TET2 gene signature, and ultimately suppressed leukemia progression in patient-derived xenografts (PDXs) [47]. Likewise, our group confirmed that long-term AA treatment prevented MN evolution in Tet2-deficient murine models [17]. However, TET2 loss due to catalytic domain lysine acetylation or missense mutations prevented this beneficial effect, which was restored by the additional use of class I and II histone deacetylase inhibitors [17]. Lower than normal AA levels have been found in patients with MN, and AA has been used at supraphysiological doses in a case of TET2 mutant AML, confirming its potential therapeutic role in TET2-mutant MN [48]. Based on these considerations, many clinical trials (NCT03682029, NCT03999723) are trying to incorporate AA in the therapeutic schemes of MN, and future data will clarify the best settings for patients suitable for this treatment option [49].
Taking into consideration the biological consequences of TET2 mutations, HMA constitutes a class of drugs currently available for patients with TET2-mutant MN. Studying a cohort of 213 MDS cases, Bejar et al. showed that TET2 mutant patients had an increased likelihood of response to HMA treatment [50]. Similarly, other studies, including ours, described a better response to HMAs in patients harboring TET2 mutations [51][52]. Indeed, as shown by competitive bone marrow transplantation experiments, HMA administration was able to significantly decrease Tet2-null cell proliferation advantage over wild-type cells (p = 0.002) [50].
Besides HMA, our group has recently developed a new therapeutic approach which entails the use of a TET-selective small-molecule inhibitor able to selectively suppress TET2-mutant cells in mouse models and TET2-mutated human leukemia xenografts while sparing normal cells [53].
Finally, in vitro and in vivo studies conducted in other models (hyperglycemic conditions) have depicted an impairment of the DNA 5-hydroxymethylome. TET2 has been identified as a substrate of the AMPK, which phosphorylates TET2 at serine 99. Increased glucose levels blocked AMPK-mediated phosphorylation at serine 99, causing the destabilization of TET2 followed by dysregulation of 5-hmC. This study also showed that administration of a biguanide (metformin) protected AMPK-mediated phosphorylation of serine 99 and increased TET2 stability and 5-hmC levels [54]. Similarly, studies conducted with AML have shown that AMPK is one of the major sensors of energy status and also for AML differentiation [55].
This entry is adapted from the peer-reviewed paper 10.3390/ijms22063135
References
- Ito, K.; Bonora, M.; Ito, K. Metabolism as master of hematopoietic stem cell fate. Int. J. Hematol. 2019, 109, 18–27.
- Kreitz, J.; Schönfeld, C.; Seibert, M.; Stolp, V.; Alshamleh, I.; Oellerich, T.; Steffen, B.; Schwalbe, H.; Schnütgen, F.; Kurrle, N.; et al. Metabolic Plasticity of Acute Myeloid Leukemia. Cells 2019, 8, 805.
- Castro, I.; Sampaio-Marques, B.; Ludovico, P. Targeting Metabolic Reprogramming in Acute Myeloid Leukemia. Cells 2019, 8, 967.
- Chapuis, N.; Poulain, L.; Birsen, R.; Tamburini, J.; Bouscary, D. Rationale for Targeting Deregulated Metabolic Pathways as a Therapeutic Strategy in Acute Myeloid Leukemia. Front. Oncol. 2019, 9.
- Saito, Y.; Chapple, R.H.; Lin, A.; Kitano, A.; Nakada, D. AMPK Protects Leukemia-Initiating Cells in Myeloid Leukemias from Metabolic Stress in the Bone Marrow. Cell Stem Cell 2015, 17, 585–596.
- Cosentino, C.; Grieco, D.; Costanzo, V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2011, 30, 546–555.
- Han, L.; Cavazos, A.; Baran, N.; Zhang, Q.; Kuruvilla, V.M.; Gay, J.P.; Feng, N.; Battula, V.L.; Kantarjian, H.M.; Daver, N.G.; et al. Mitochondrial Oxphos as Survival Mechanism of Minimal Residual AML Cells after Induction Chemotherapy: Survival Benefit by Complex I Inhibition with Iacs-010759. Blood 2019, 134, 5161.
- Jacque, N.; Ronchetti, A.M.; Larrue, C.; Meunier, G.; Birsen, R.; Willems, L.; Saland, E.; Decroocq, J.; Maciel, T.T.; Lambert, M.; et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015, 126, 1346–1356.
- DiNardo, C.D.; Cortes, J.E. Mutations in AML: Prognostic and therapeutic implications. Hematol. Am. Soc. Hematol. Educ. Progr. 2016, 2016, 348–355.
- Bassal, M.A.; Leo, P.; Samaraweera, S.E.; Maung, K.Z.Y.; Babic, M.; Venugopal, P.; Cheah, J.; Hahn, C.N.; Scott, H.S.; Glazov, E.; et al. Metabolic Profiling of Adult Acute Myeloid Leukemia (AML). Blood 2016, 128, 1684.
- Chiba, S. Dysregulation of TET2 in hematologic malignancies. Int. J. Hematol. 2017, 105, 17–22.
- Lorsbach, R.B.; Moore, J.; Mathew, S.; Raimondi, S.C.; Mukatira, S.T.; Downing, J.R. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia 2003, 17, 637–641.
- Ono, R.; Taki, T.; Taketani, T.; Taniwaki, M.; Kobayashi, H.; Hayashi, Y. LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10;11)(q22;q23). Cancer Res. 2002, 62, 4075–4080.
- Ko, M.; Huang, Y.; Jankowska, A.M.; Pape, U.J.; Tahiliani, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Koh, K.P.; Ganetzky, R.; et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010, 468, 839–843.
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307.
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479.
- Guan, Y.; Greenberg, E.F.; Hasipek, M.; Chen, S.; Liu, X.; Kerr, C.M.; Gackowski, D.; Zarakowska, E.; Radivoyevitch, T.; Gu, X.; et al. Context dependent effects of ascorbic acid treatment in TET2 mutant myeloid neoplasia. Commun. Biol. 2020, 3, 493.
- Bochtler, M.; Kolano, A.; Xu, G.L. DNA demethylation pathways: Additional players and regulators. BioEssays 2017, 39, 1–13.
- Verma, N.; Pan, H.; Doré, L.C.; Shukla, A.; Li, Q.V.; Pelham-Webb, B.; Teijeiro, V.; González, F.; Krivtsov, A.; Chang, C.J.; et al. TET proteins safeguard bivalent promoters from de novo methylation in human embryonic stem cells. Nat. Genet 2018, 50, 83–95.
- Zheng, G.; Fu, Y.; He, C. Nucleic acid oxidation in DNA damage repair and epigenetics. Chem. Rev. 2014, 114, 4602–4620.
- Dawlaty, M.M.; Breiling, A.; Le, T.; Barrasa, M.I.; Raddatz, G.; Gao, Q.; Powell, B.E.; Cheng, A.W.; Faull, K.F.; Lyko, F.; et al. Loss of Tet enzymes compromises proper differentiation of embryonic stem cells. Dev. Cell 2014, 29, 102–111.
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133.
- Quivoron, C.; Couronné, L.; Della Valle, V.; Lopez, C.K.; Plo, I.; Wagner-Ballon, O.; Do Cruzeiro, M.; Delhommeau, F.; Arnulf, B.; Stern, M.H.; et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 2011, 20, 25–38.
- Dawlaty, M.M.; Breiling, A.; Le, T.; Raddatz, G.; Barrasa, M.I.; Cheng, A.W.; Gao, Q.; Powell, B.E.; Li, Z.; Xu, M.; et al. Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev. Cell 2013, 24, 310–323.
- Secardin, L.; Limia, C.E.G.; di Stefano, A.; Bonamino, M.H.; Saliba, J.; Kataoka, K.; Rehen, S.K.; Raslova, H.; Marty, C.; Ogawa, S.; et al. TET2 haploinsufficiency alters reprogramming into induced pluripotent stem cells. Stem Cell Res. 2020, 44, 101755.
- Guan, Y.; Hasipek, M.; Tiwari, A.D.; Maciejewski, J.P.; Jha, B.K. TET-dioxygenase deficiency in oncogenesis and its targeting for tumor-selective therapeutics. Semin. Hematol. 2021, 58, 27–34.
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221.
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247.
- Coltro, G.; Mangaonkar, A.A.; Lasho, T.L.; Finke, C.M.; Pophali, P.; Carr, R.; Gangat, N.; Binder, M.; Pardanani, A.; Fernandez-Zapico, M.; et al. Clinical, molecular, and prognostic correlates of number, type, and functional localization of TET2 mutations in chronic myelomonocytic leukemia (CMML)-a study of 1084 patients. Leukemia 2020, 34, 1407–1421.
- Hirsch, C.M.; Nazha, A.; Kneen, K.; Abazeed, M.E.; Meggendorfer, M.; Przychodzen, B.P.; Nadarajah, N.; Adema, V.; Nagata, Y.; Goyal, A.; et al. Consequences of mutant TET2 on clonality and subclonal hierarchy. Leukemia 2018, 32, 1751–1761.
- Gurnari, C.; Fabiani, E.; Falconi, G.; Travaglini, S.; Ottone, T.; Cristiano, A.; Voso, M.T. From Clonal Hematopoiesis to Therapy-Related Myeloid Neoplasms: The Silent Way of Cancer Progression. Biology 2021, 10, 128.
- Pan, F.; Wingo, T.S.; Zhao, Z.; Gao, R.; Makishima, H.; Qu, G.; Lin, L.; Yu, M.; Ortega, J.R.; Wang, J.; et al. Tet2 loss leads to hypermutagenicity in haematopoietic stem/progenitor cells. Nat. Commun. 2017, 8, 15102.
- Awada, H.; Nagata, Y.; Goyal, A.; Asad, M.F.; Patel, B.; Hirsch, C.M.; Kuzmanovic, T.; Guan, Y.; Przychodzen, B.P.; Aly, M.; et al. Invariant phenotype and molecular association of biallelic TET2 mutant myeloid neoplasia. Blood Adv. 2019, 3, 339–349.
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011, 20, 11–24.
- Li, Z.; Cai, X.; Cai, C.L.; Wang, J.; Zhang, W.; Petersen, B.E.; Yang, F.C.; Xu, M. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 2011, 118, 4509–4518.
- Pronier, E.; Almire, C.; Mokrani, H.; Vasanthakumar, A.; Simon, A.; da Costa Reis Monte Mor, B.; Massé, A.; Le Couédic, J.P.; Pendino, F.; Carbonne, B.; et al. Inhibition of TET2-mediated conversion of 5-methylcytosine to 5-hydroxymethylcytosine disturbs erythroid and granulomonocytic differentiation of human hematopoietic progenitors. Blood 2011, 118, 2551–2555.
- Zhang, Y.W.; Wang, Z.; Xie, W.; Cai, Y.; Xia, L.; Easwaran, H.; Luo, J.; Yen, R.C.; Li, Y.; Baylin, S.B. Acetylation Enhances TET2 Function in Protecting against Abnormal DNA Methylation during Oxidative Stress. Mol. Cell 2017, 65, 323–335.
- Scopim-Ribeiro, R.; Machado-Neto, J.A.; Campos Pde, M.; Silva, C.A.; Favaro, P.; Lorand-Metze, I.; Costa, F.F.; Saad, S.T.; Traina, F. Ten-eleven-translocation 2 (TET2) is downregulated in myelodysplastic syndromes. Eur. J. Haematol. 2015, 94, 413–418.
- Scopim-Ribeiro, R.; Machado-Neto, J.A.; de Melo Campos, P.; Niemann, F.S.; Lorand-Metze, I.; Costa, F.F.; Olalla Saad, S.T.; Traina, F. Low Ten-eleven-translocation 2 (TET2) transcript level is independent of TET2 mutation in patients with myeloid neoplasms. Diagn. Pathol. 2016, 11, 28.
- Jankowska, A.M.; Szpurka, H.; Tiu, R.V.; Makishima, H.; Afable, M.; Huh, J.; O’Keefe, C.L.; Ganetzky, R.; McDevitt, M.A.; Maciejewski, J.P. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood 2009, 113, 6403–6410.
- Coutinho, D.F.; Monte-Mór, B.C.; Vianna, D.T.; Rouxinol, S.T.; Batalha, A.B.; Bueno, A.P.; Boulhosa, A.M.; Fernandez, T.S.; Pombo-de-Oliveira, M.S.; Gutiyama, L.M.; et al. TET2 expression level and 5-hydroxymethylcytosine are decreased in refractory cytopenia of childhood. Leuk. Res. 2015, 39, 1103–1108.
- Zhang, T.J.; Zhou, J.D.; Yang, D.Q.; Wang, Y.X.; Wen, X.M.; Guo, H.; Yang, L.; Lian, X.Y.; Lin, J.; Qian, J. TET2 expression is a potential prognostic and predictive biomarker in cytogenetically normal acute myeloid leukemia. J. Cell. Physiol. 2018, 233, 5838–5846.
- Wang, J.; Li, F.; Ma, Z.; Yu, M.; Guo, Q.; Huang, J.; Yu, W.; Wang, Y.; Jin, J. High Expression of TET1 Predicts Poor Survival in Cytogenetically Normal Acute Myeloid Leukemia from Two Cohorts. EBioMedicine 2018, 28, 90–96.
- Zhang, T.; Zhao, Y.; Zhao, Y.; Zhou, J. Expression and prognosis analysis of TET family in acute myeloid leukemia. Aging 2020, 12, 5031–5047.
- Peng, D.; Ge, G.; Gong, Y.; Zhan, Y.; He, S.; Guan, B.; Li, Y.; Xu, Z.; Hao, H.; He, Z.; et al. Vitamin C increases 5-hydroxymethylcytosine level and inhibits the growth of bladder cancer. Clin. Epigen. 2018, 10, 94.
- Agathocleous, M.; Meacham, C.E.; Burgess, R.J.; Piskounova, E.; Zhao, Z.; Crane, G.M.; Cowin, B.L.; Bruner, E.; Murphy, M.M.; Chen, W.; et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 2017, 549, 476–481.
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095.e20.
- Das, A.B.; Kakadia, P.M.; Wojcik, D.; Pemberton, L.; Browett, P.J.; Bohlander, S.K.; Vissers, M.C.M. Clinical remission following ascorbate treatment in a case of acute myeloid leukemia with mutations in TET2 and WT1. Blood Cancer J. 2019, 9, 82.
- Aldoss, I.; Mark, L.; Vrona, J.; Ramezani, L.; Weitz, I.; Mohrbacher, A.M.; Douer, D. Adding ascorbic acid to arsenic trioxide produces limited benefit in patients with acute myeloid leukemia excluding acute promyelocytic leukemia. Ann. Hematol. 2014, 93, 1839–1843.
- Bejar, R.; Lord, A.; Stevenson, K.; Bar-Natan, M.; Pérez-Ladaga, A.; Zaneveld, J.; Wang, H.; Caughey, B.; Stojanov, P.; Getz, G.; et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood 2014, 124, 2705–2712.
- Itzykson, R.; Kosmider, O.; Cluzeau, T.; Mansat-De Mas, V.; Dreyfus, F.; Beyne-Rauzy, O.; Quesnel, B.; Vey, N.; Gelsi-Boyer, V.; Raynaud, S.; et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia 2011, 25, 1147–1152.
- Traina, F.; Visconte, V.; Elson, P.; Tabarroki, A.; Jankowska, A.M.; Hasrouni, E.; Sugimoto, Y.; Szpurka, H.; Makishima, H.; O’Keefe, C.L.; et al. Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia 2014, 28, 78–87.
- Guan, Y.; Tiwari, A.D.; Phillips, J.G.; Hasipek, M.; Grabowski, D.R.; Pagliuca, S.; Gopal, P.; Kerr, C.M.; Adema, V.; Radivoyevitch, T.; et al. A Therapeutic Strategy for Preferential Targeting of TET2-Mutant and TET Dioxygenase–Deficient Cells in Myeloid Neoplasms. Blood Cancer Discov. 2020.
- Wu, D.; Hu, D.; Chen, H.; Shi, G.; Fetahu, I.S.; Wu, F.; Rabidou, K.; Fang, R.; Tan, L.; Xu, S.; et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature 2018, 559, 637–641.
- Jacquel, A.; Luciano, F.; Robert, G.; Auberger, P. Implication and Regulation of AMPK during Physiological and Pathological Myeloid Differentiation. Int. J. Mol. Sci. 2018, 19, 2991.