The high prevalence of type 2 diabetes mellitus (T2DM), together with the fact that current treatments are only palliative and do not avoid major secondary complications, reveals the need for novel approaches to treat the cause of this disease. Efforts are currently underway to identify therapeutic targets implicated in either the regeneration or re-differentiation of a functional pancreatic islet β-cell mass to restore insulin levels and normoglycemia. However, T2DM is not only caused by failures in β-cells but also by dysfunctions in the central nervous system (CNS), especially in the hypothalamus and brainstem.
- metabesity
- T2DM
- obesity
- inflammation
- pancreatic islet
- astrocytes
1. Introduction
Diabetes mellitus (DM) is currently a major social and economic burden worldwide. The global epidemic proportion of DM is one of the most important health problems of the 21st century, being the cause of four million deaths in 2017 [1]. According to the International Diabetes Federation, 425 million people were affected by DM in 2017 (8.8% worldwide prevalence), and this number is expected to rise to 629 million people by 2045 [1]. This alarming increase in the incidence of DM is mainly due to the increase in the number of type 2 DM (T2DM) sufferers, which accounts for 90–95% of all DM. T2DM is associated with environmental and nutritional factors, as well as lifestyle, operating on a genetic susceptibility background. Approximately 80% of T2DM patients are obese, and the increase in the global epidemic of obesity explains the dramatic explosion of T2DM incidence over the past two decades [1]. Over nutrition-derived metabolic dysfunctions, henceforth denoted as metabesity, contributes to the apparition of a chronic low-grade inflammation [2,3], which is recognized as the pathogenic link with T2DM. This inflammation initially targets peripheral tissues specialized in metabolism such as liver, adipose tissue, and pancreatic islets, and it plays a key role in insulin resistance and β-cell dysfunction [4,5,6]. Inflammation also affects the central nervous system (CNS), likely being one of the causes underlying the increased incidence of neurodegenerative diseases among T2DM patients. Of note, hypothalamic inflammation is arising as a key pathophysiological process in metabesity, linked to hypothalamic leptin and insulin resistance [7,8,9].
T2DM is a progressive metabolic disorder classically defined by chronic hyperglycemia due to the combination of an abnormal insulin secretion by pancreatic islet β-cells and increased insulin resistance of insulin-target tissues (adipose tissue, skeletal muscle, liver, and brain) [10]. Guidelines for T2DM management include, as first-line therapy, serious lifestyle interventions such as physical exercise, while long-term add-on therapies include medication in order to increase insulin secretion and sensitivity [11]. Although these treatments are adequate to improve hyperglycemia, they alleviate symptoms rather than target the root-cause of the disease and lead to the development of secondary complications [12]. For example, sulfonylureas that impel insulin secretion were shown to cause β-cell death and patients are thenceforth confined to daily insulin injections to control glucose homeostasis [13]. However, new classes of pharmacological agents such as sodium glucose transporter 2 SGLT2 inhibitors and glucagon-like peptide-1 (GLP-1) receptor agonists significantly improve the prognosis of T2DM patients [14]. Nonetheless, a better understanding of T2DM etiology is mandatory to develop more effective therapies to avoid long-term complications and early death associated with this disease, and to restrain the apparition of neurodegenerative disorders such as Alzheimer’s disease, dubbed T3DM [15]. In recent years, the CNS gained much interest as a key regulator of glucose/energy homeostasis [16]. Thus, T2DM is not only caused by failures in pancreatic β-cells but also dysfunctions in the CNS that could lead to the development of this disease. Moreover, metabesity-associated chronic inflammation also affects the CNS-mediated control of blood glucose levels, exacerbating the disease. Herein, we review the evidence that supports a main role for the CNS in glucose homeostasis, with a special focus on astrocytes, and how interventions that simultaneously target both the CNS and pancreatic islets may synergistically improve the regulation of blood glucose levels and metabolism in T2DM patients.
2. Cell Types of the Hypothalamus Implicated in Energy Homeostasis
2.1. Hypothalamic Neurons
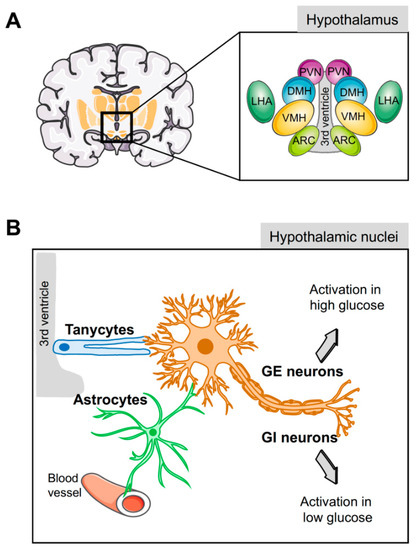
2.2. Hypothalamic Glial Cells
Virchow in 1846 initially coined the term neuroglia to describe a connective substance embedding neurons in the brain. Subsequently, the Spanish neuroscientist Ramón y Cajal reported on glial cells and their role in brain physiology [106]. The direct contact of glial cells to blood vessels and glucose sensing neurons raises the possibility that these cells mediate and bridge glucose signaling [105,107,108]. Indeed, the hypothalamic astrocytes and tanycytes, two types of glial cells, participate in the regulation of glucose and energy homeostasis [17,104,107,109] by modulating both peripheral and central glucose levels [60] and providing energy substrates to neighbouring neurons.
Tanycytes are specialised ependymoglial cells surrounding the lateral walls and floor of the third ventricle [110]. Cell bodies of tanycytes are in direct contact with the cerebral spinal fluid (CSF) and have a long process projected into the hypothalamus, a privileged position to sense hormones and nutritional signals from either the periphery or the CSF and relay them to the brain (Figure 1B) [110]. Astrocytes also have a strategic physiological location in close proximity to the blood–brain barrier (BBB) and, with their end-feet covering the surface of capillaries, support the hypothesis that astrocytes supply neuron energetic demands in a process known as the astrocyte–neuron lactate shuttle (Figure 1B) [107,111]. In the absence of glucose, the lactate derived from astrocytic glycogen metabolism maintains neuronal function [112].
Recent evidence highlights the important role of these two hypothalamic glial cells in glucose sensing and glucose homeostasis. Central icv injection of fibroblast growth factor 1 (FGF1) induces diabetes remission in animal models of T2DM in a process mediated by tanycyte activation that enhances insulin-independent glucose clearance [56,113]. Noteworthy, this effect of FGF1 administration is accompanied by a preservation of β-cell function [113]. Additionally, image analyses of rat brain slices showed that, in response to glucose, tanycytes display an increase in intracellular Ca2+ stimulating glycolysis and lactate release [110,114,115,116,117]. This lactate is then relayed to POMC neurons in the ARC triggering exocytosis of αMSH to produce satiety [118]. This mechanism is dependent on the GLUT-2, as inhibition of this transporter specifically in tanycytes disrupts the hypothalamic glucosensing mechanism, reducing glucose uptake and lactate production. Consequently, POMC neuronal activity is interrupted and the control of feeding behavior is altered [119]. Noteworthy, the astrocytic glucose transporters have also a critical role in glucosensing. As an example of this, the re-expression of Glut2 in astrocytes of Glut2 null mice restores glucagon secretion in response to hypoglycemia [120,121], indicating that hypothalamic astrocytes via GLUT-2 are a key part of the central glucose sensing machinery and play a key role in the regulation of glycemia. Likewise, repression of hypothalamic GLUT-1 levels due to sustained hyperglycemia blunted the decrease in systemic glucose production, whereas GLUT-1 over-expression in hypothalamic astrocytes restored glucose-sensing capacity of these cells [122]. Nevertheless, glucose transporters are not the only mechanism used by these glial cells to sense glucose. The fact that tanycytes also respond to non-metabolizable glucose analogs, 2-deoxyglucose and methyl glucopyranoside [115,117], suggests that additional glucosensing mechanisms are implicated. In this regard, G-protein-coupled receptors (GPCRs) were proposed to be implicated in glucose sensing and regulation [123]. Interestingly, the GPCR cannabinoid CB1 receptor, a well-known player in energy homeostasis, is expressed in astrocytes and facilitates neuron–astrocyte communication [124]. Although the role of the CB1 receptor in glucose sensing and homeostasis is still unknown, some evidence points to the regulation of the leptin signaling pathway and glycogen storage in astrocytes [125].
3. Brain/Islet Glucose Homeostasis Axis Orchestrated by HMG20A?
Glucose homeostasis is maintained through a network of different organs and tissues that respond to alterations in glucose levels in an organized manner orchestrated by the brain. As described here, the hypothalamus senses glucose fluctuations and activates neuronal circuits that contribute to the regulation of insulin and glucagon secretion by pancreatic endocrine cells, as well as glucose metabolic pathways in liver, fat, and muscle, altogether resulting in the maintenance of glucose homeostasis. Nonetheless, a centralized model of glucosensing relies, as a last resort, on the adequate function of the pancreatic islets; thus, a brain–islet axis is indispensable for the fine-tuning of glucose homeostasis. Interestingly, both pancreatic β-cell and CNS cells possess similar developmental genetic programs that include common key transcription factors such as NEUROD and ISL1, as well as sharing identical regulated exocytosis machinery [23,182,183]. As such, potential epigenetic master regulators could foreseeably orchestrate both tissues/organs in cooperatively controlling glucose homeostasis. We and others showed that HMG20A is involved in neuronal and β-cell mature function [41,184]. As HMG20A regulates the expression of key genes such as NEUROD, GK, and GLUTs common to both astrocytes and β-cells, as well as genes such as insulin and GFAP restricted to each cell type, it is tempting to speculate that this chromatin remodeling factor is a common master regulator in β-cells and astrocytes, integrating inputs to altered glucose levels and stressful physiological conditions that can influence glucose metabolism (Figure 2). This premise is currently under investigation.
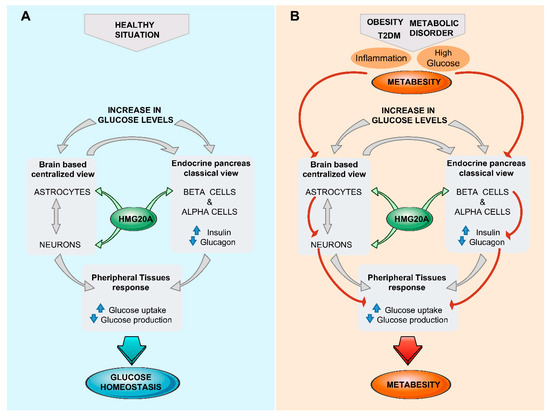
Figure 2. The brain (astrocyte)/islet axis in metabesity. (A) In response to increased glucose levels, a dialogue between brain astrocytes/neurons and islets coordinates glucose homeostasis by peripheral tissue relaying. The chromatin-remodeling factor HMG20A is a common denominator expressed in both astrocytes and islets that facilitates integration of input signals. (B) In the advent of metabesity, characterized by obesity, insulin resistance, and inflammation, which alter the expression of HMG20A, the brain/islet axis is short-circuited, precipitating the disease state. T2DM, type 2 diabetes mellitius.
This entry is adapted from the peer-reviewed paper 10.3390/genes10050350