G protein-coupled receptors (GPCRs) are the most important regulators of cardiac function and are commonly targeted for medical therapeutics. Formyl-Peptide Receptors (FPRs) belong to the GPCR superfamily and include three members (FPR1, FPR2 and FPR3). FPRs are functionally expressed in several cells and tissues where they can significantly contribute to inflammatory disorders, cancer, infections and cardiovascular pathologies. FPRs stimulation induces phosphorylation of several signaling proteins modulating different cellular functions such as cell growth, proliferation, intracellular communication, migration, differentiation, apoptosis, and survival. FPRs can also modulate oxidative stress through nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-dependent reactive oxygen species (ROS) production whose dysregulation has been observed in different cardiovascular diseases.
- formyl-peptide receptors
- NADPH oxidase
- reactive oxygen species
- annexin A1
- lipoxin A4
- serum-amyloid alpha
1. Introduction
The most important regulators of cardiac function, such as contractility, remodeling and heart rate, belong to the superfamily of G protein-coupled receptors (GPCRs) [1]. GPCRs represent the largest and the most versatile family of cell surface receptors, and since they are involved in a wide variety of physio-pathological processes they are commonly targeted for medical therapies [2]. Formyl-Peptide Receptors (FPRs) belong to the sensitive pertussis toxin-GPCR superfamily and play an emerging role in cardiovascular pathologies [3,4,5,6,7], neuronal diseases [8,9] and cancer progression [10,11,12]. FPRs family consist of three members (FPR1, FPR2 and FPR3) widely expressed in several tissues and cell types, where they modulate several biological functions, such as angiogenesis, cell proliferation and protection against cell death [9].
Initially discovered as the receptor for the formylated bacterial product N-formyl-methionine-leucyl-phenylalanine (N-fMLF), FPR1 is highly expressed in cells of bone marrow and immune system, but also in lungs, brain, and gastrointestinal tract cells [13,14]. FPR2 is the most promiscuous and widespread receptor in the FPR family. It is mainly expressed in cells of the bone marrow, immune system, gastrointestinal tract, female organ tissues, endocrine glands, brain, liver, gallbladder, and pancreas [13,14]. FPR2 is also functionally expressed on the nuclear membrane since it shows a nuclear localization sequence in the third cytoplasmic loop [15].
The biological role of FPR3 is not completely known, which is mainly expressed in monocytes and dendritic cells but not in neutrophils. It is not localized on the cell surface, like its counterpart receptors, but it resides in intracellular vesicles [13]. Most FPR ligands induce cell chemotaxis, calcium flux and phagocytosis. Some FPR agonists elicit inflammatory processes while other ligands activate proresolving or anti-inflammatory pathways, depending on the nature of the ligands. In general, bacterial and mitochondrial formylated peptides activate a proinflammatory cell response, while Annexin A1 (ANXA1) and Lipoxin A4 (LXA4) are anti-inflammatory FPR2 ligands [16]. The switch between FPR2-mediated pro- and anti-inflammatory cell responses depends on conformational changes of the receptor due to ligand binding; anti-inflammatory ligands such as ANXA1 induce the formation of FPR homodimers and, in turn, the release of inflammation-resolving cytokines. On the other hand, inflammatory ligands such as serum-amyloid alpha (SAA) do not cause receptor homodimerization [12,17].
ANXA1 is a 37-kDa member of a superfamily of 13 annexin proteins widely expressed in cells of myeloid origin [18]. The proresolving effects of this protein are mediated by the L-selectin shedding resulting in reduced neutrophil adhesion to the endothelium and limited transmigration [18].
Therefore, FPRs can be stimulated by a plethora of endogenous or exogenous ligands, with different chemical properties [12,19], suggesting that they can be considered pattern recognition receptors able to recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular pattern (DAMPs) [20,21]. Both PAMPs and DAMPs cause a systemic inflammatory response syndrome by activating innate immunity [12]. DAMPs include proteins, nucleic acids, extracellular matrix components, and lipid mediators released from necrotic cells to guide neutrophils to sites of sterile inflammation. DAMPs take part in the early reaction after stroke onset and contribute to the acute conditions that result from sterile inflammation, also in ischemia-reperfusion injury (IRI) [22]. Among DAMPs, mitochondrial formylated peptides released from necrotic or damaged cells can interact with members of the FPR family triggering neutrophil-mediated organ injury [21]. Furthermore, mitochondrial dysfunctions contribute to sterile inflammation, by releasing mitochondrial N-formyl peptides that can bind FPRs [23], in a variety of cardiac diseases including coronary artery disease, left ventricular hypertrophy and heart failure [24,25,26].
An increased risk of cardiac injury correlates with systemic inflammation such as in chronic obstructive pulmonary disease (COPD)-associated systemic inflammation [27,28]. Indeed, hypoxemia observed in patients affected by COPD can lead to pulmonary vasoconstriction and vascular remodeling, resulting in right-ventricular diastolic dysfunction [29]. Moreover, hypoxemia in patients with COPD can be also related to an impaired cardiac repolarization by increasing the risk of ventricular arrhythmias and sudden cardiac death [29,30]. FPRs contribute to the COPD progression. FPR1 is involved in impaired epithelial repair responses associated with cigarette smoke, which is considered one of the leading causes of COPD [31]. Moreover, decreased FPR2 and FPR3, as well as defective ANXA1 generation, were observed in COPD patients [32].
Other FPR ligands, such as ANXA1 and SAA, are involved in cardiovascular disease progression. In fact, ANXA1 and its N-terminal peptides (Ac2-26 and Ac9-25) are endogenous anti-inflammatory and proresolving mediators, known to have significant effects in resolving inflammation in a variety of disease models and showing therapeutic potential in IRI [33]. ANXA1 has also protective role in myocardial infarction (MI) and stroke [34]. SAA is a sensitive marker of an acute inflammatory state [35] and although it is mainly associated with amyloidosis, it is involved in early atherogenic processes [36] and represents a marker of cardiovascular events [37,38].
Reactive oxygen species (ROS) are critical signaling molecules involved in both pathological and physiological cardiovascular processes. Several cytosolic sources contribute to the intracellular ROS pool, such as NADPH oxidase (NOX), xanthine oxidase, cyclooxygenases, and cytochrome P450 enzymes. Mitochondrial sources of ROS include the respiratory chain, monoamine oxidases, p66shc, and NOX4 [39]. Elevated levels of ROS result in oxidative stress and damage to DNA, lipids and proteins, as well as in impairing mitochondrial functions, thus contributing to cell death. Dysregulation of ROS generation and consequent oxidative stress has been observed in different cardiovascular diseases, including cardiac hypertrophy [40], heart failure [41], cardiac IRI [42], diabetic cardiomyopathy [43], and cardiomyopathy associated with Duchenne/Becker muscular dystrophy [44].
Since ROS play a crucial role in cardiovascular physiopathological processes, several studies are focused on identifying the molecular mechanisms of regulation of ROS production, as well as on developing therapies to counteract ROS production in heart diseases. In leukocytes and polymorphonucleate cells (PMNs) FPRs modulate oxidative burst through NOX-dependent ROS generation [45]. Indeed, FPRs stimulation triggers multiple phosphorylations of intracellular signaling molecules, such as extracellular signal-related kinases 1/2 (ERKs), protein kinase C (PKC), protein kinase B (PKB), mitogen-activated protein (MAP) kinases p38 (p38MAPK), phosphatidylinositol-3-kinase (PI3K), and phospholipase C (PLC), as well as nonsignaling proteins, such as p47phox and p67phox, required for NOX activation [45,46]. In other cell types, NOX-dependent ROS generation triggered by FPR1 and FPR2 stimulation is necessary to modulate several intracellular redox signaling pathways [47]. In fact, ROS can act as second messengers, by inhibiting protein tyrosine phosphatase (PTPs) activity and, in turn, promoting the phosphorylated state of protein tyrosine kinases (PTKs), thus regulating intracellular signaling cascades [48]. In support of these comments, a phospho-proteomic analysis on FPR2-stimulated cells identified 290 differentially phosphorylated proteins, including kinases and phosphatases, as new phosphorylation sites which, at least in part, are regulated by NOX-mediated ROS generation [14,45,46]. Furthermore, NOX-dependent transactivation of several Tyrosine Kinase Receptors (TKRs) by FPR1 or FPR2 stimulation [48] has been described in epithelial [49,50], neuronal [51] and endothelial cells [52].
2. Formyl-Peptide Receptor 1
FPR1 shows the highest binding affinity for N-formylated peptides among all members of the FPRs family. Its stimulation results in PI3K, PKC, p38MAPK, and ERKs activation, as well as in increase of chemotaxis and NOX-dependent ROS production [53,54]. Both FPR1 and FPR2 cooperate with other GPCRs to modulate chemotaxis [10], as demonstrated by the observation that neutrophil accumulation in tissue injury is controlled by multiple chemoattractants, such as IL-8 (CXCL8), CXCL7 and CXCL1 [10]. However, FPR1 and FPR2 act as the first players in sensing chemotaxis signals resulting in rapid neutrophil infiltration [55]. In fact, their activation by the agonists produced at the injury site triggers a signaling cascade that culminates in the neutrophil migration, increased phagocytosis and superoxide release.
FPR1 is also expressed in several tumors. In highly malignant glioblastoma multiforme (GBM) cells it responds to the endogenous chemotactic ligand ANXA1 released by necrotic GBM cells [56]. In these cells, activated FPR1 enhances the survival, invasiveness, and production of angiogenic factors by cooperating with the epidermal growth factor receptor [56,57]. FPR1 is implicated in the production of angiogenic factors also in human liver cancer cells, where it promotes cell invasion and proliferation [58]. In human breast cancer cells, FPR1 and FPR2 interact with ANXA1 to enhance tumor cell proliferation [59]. Furthermore, in human gastric cancer (GC), FPR1 levels are correlated with more aggressive submucosal and serosal invasion accompanied by worse outcomes in patients [60]. Interestingly, FPR1 silencing significantly enhances GC tumorigenicity, suggesting that this receptor exerts a tumor suppressor function by inhibiting angiogenesis [57].
FPR1 contributes to endothelial/cardiac/cerebral dysfunction and repair in intermittent hypoxia/reoxygenation (IHR)/IRI [61,62,63], which is a common feature of several diseases, such as stroke and MI. Ischemia refers to a decrease in blood flow. Reperfusion injury is instead associated with initial blood-borne neutrophil infiltration, giving rise to an inflammatory response that results in tissue injury. In fact, the damaged tissue displays important signs of inflammation and microvascular injury that, unless resolved, lead to tissue dysfunction. Restoration of blood flow to a previously ischemic region is essential to prevent irreversible tissue damage but it is not always beneficial. During reperfusion, a great amount of damage occurs to the tissue, although a significant level of injury occurs due to ischemia itself. Several events are involved in the inflammatory cascade following myocardial injury, including leukocyte activation, lipid peroxidation and an increase of vascular permeability [64]. Leukocyte recruitment occurs in the microvasculature and involves a complex multistep cascade [63]. FPR1 gene deficiency reduces the risk of heart injury induced by IRI [65], as demonstrated by a significant decrease of collagen volume fraction, infarct area and apoptotic index observed in the heart of FPR1-silenced mice. This correlates with a reduction of p38MAPK, ERKs, jun kinase (JNK), MMP-2, TIMP-2, NF-kB, Bax, phospho-p38MAPK, phospho-ERKs, phospho-JNK levels, and cell apoptosis rate, as well as with an enhanced Bcl-2 level, cell proliferation and cell cycle progression. Therefore, these observations suggest that FPR1silencing reduces inflammation, cardiomyocyte apoptosis and ventricular remodeling in IRI, through the suppression of the MAPK signaling pathway [66]. Consistently, FPR1 expression levels are higher in patients with cardiac IRI [66].
Blockade of FPR1 functions in preventing IRI has been observed also in the liver. Mitochondrial DAMPs, including formyl peptides, are recognized by FPR1 [21] and the signaling triggered by this receptor is responsible for regulating neutrophil chemotaxis, which allows their migration into the necrotic area in hepatic ischemia-reperfusion [67]. Pretreatment with cyclosporine H (CSH), a selective FPR1 antagonist, prevents hepatic IRI as demonstrated by decreased serum transaminase and inflammatory cytokine levels, reduced hepatocyte necrosis/apoptosis and oxidative stress in a mouse model. Moreover, FPR1 blockade also inhibits the accumulation of neutrophils in the necrotic area [67].
IRI occurs in a sterile environment and involves acute inflammation and innate immune cell activation, leading to rapid infiltration of neutrophils. Therefore, inflammatory responses, that crucially contribute to reperfusion injury, are, at least in part, associated with circulating neutrophils activation. FPR1 plays an important role in neutrophil function and its expression is rapidly upregulated in response to inflammatory stimuli, which contribute to tissue damage through several mechanisms: (i) FPR1-mediated NOX-dependent ROS generation; (ii) release of elastases, cathepsin G, proteinase and other proteolytic enzymes; (iii) release of cytokines from local cells and subsequent neutrophil recruitment; (iv) obstruction of capillaries by neutrophils thus contributing to the no-flow phenomenon [68]. Therefore, FPR1 and the neutrophil-mediated inflammatory cascade during reperfusion represent an important target for therapeutic intervention.
Moreover, development of resistant to degradation FPR agonists as potential therapeutics could be useful for a range of inflammatory disorders. In particular, Cmpd17b is a small biased FPR1/FPR2 agonist that significantly reduces necrosis in cardiomyocytes subjected to hypoxia–reoxygenation [69].
ANXA1 is expressed constitutively in many cells, including neutrophils [68]. It is released upon neutrophil adhesion to endothelial cells, and it binds to FPRs receptors to suppress inflammatory signaling cascades [68]. Exogenous administration of ANXA1 exerts protective and anti-inflammatory actions and acts as a second messenger for many glucocorticoid effects in neutrophils, macrophages and other circulating inflammatory cells. Many N-terminal ANXA1-derived peptides, such as Ac2-26, display similar activity to the full-length protein, showing potent inhibition of neutrophil function and thus representing cardioprotective factors against myocardial IRI [68]. In particular, Ac2-26 preserves inotropic responsiveness at the level of ventricular muscle and contractile function of cardiac muscle in vitro [70,71,72,73]. The anti-inflammatory properties of ANXA1-derived peptides have been largely attributed to their anti-neutrophil actions in vivo and to activation of members of the FPR family [70]. ANXA1 affects myocardial function and is an endogenous regulator of post-ischemic recovery of left ventricular (LV) function. Ac2-26 rescues LV function on reperfusion, via activation of FPR1. Therefore, ANXA1-based therapies may represent a novel clinical approach for the prevention and treatment of myocardial reperfusion injury [74].
FPR1 is a potential biomarker for acute MI. In fact, it has been identified, together with genes involved in inflammation, as a differentially expressed gene in human MI blood tissues, compared with normal blood tissues [75,76]. In addition, FPR2 activation is involved in cardiac repair after MI. This suggests that the deregulated FPRs expression in blood tissue might account for the occurrence of acute MI [62,76]. Thus, FPRs represent novel therapeutic targets in MI, where inflammation is a major contributing mechanism. Cardiomyocyte survival and preservation of LV function seem to be enhanced by FPR1 [74], whereas FPR2 is responsible for attenuation of inflammation [77]. Therefore, dual FPR1/FPR2 agonists may be useful for lessening MI injury [69].
The spleen contributes to inflammatory responses during cardiac remodeling after MI, as demonstrated by the observation that the cardio-splenic axis plays a pivotal role in exacerbating MI size during post-ischemic reperfusion [78]. Interestingly, FPR1 blockade prevents the activation of this cardio-splenic signaling axis and abrogates the reperfusion-induced exacerbation of infarct size. In fact, myocardial neutrophil infiltration is reduced by cFLFLF, a specific FPR1 antagonist, which abrogates the infarct-exacerbating effect in mice. Consistently, in mice sham spleens the injection of ischemic heart homogenate significantly increases FPR1 expression [78].
Multiple genetic determinants and environmental factors contribute to modulate blood pressure (BP) levels [79]. Genome-wide analysis studies have identified candidate loci on the long arm of chromosome 19 (19q) harbors linked to increased BP levels and related pathological phenotypes [80,81]. FPR1 gene localizes in position 19q 13.3 and seems to play a key role in the physiopathology of hypertension, being crucially involved in the modulation of inflammation and in BP related metabolic pathways [82]. Polymorphisms of FPR1 are associated with inflammation and BP. In particular, FPR1 C32T (rs5030878), a nonsynonymous coding single nuclear polymorphism (SNP)(Ile11Thr), results associated with increased C-reactive protein (CRP) levels linearly related to BP [83,84,85].
Abdominal aortic aneurysm (AAA) is characterized by a progressive aortic dilatation and weakening of the vascular wall that may provoke an aortic rupture, which often is fatal. Pathogenesis of AAA includes extracellular matrix degradation, vascular smooth muscle cell de-differentiation/apoptosis, ROS accumulation, and inflammatory cell infiltration [86,87]. PMNs infiltration and activation, and chemokine-mediated neutrophil recruitment contribute to the pathogenesis of AAA and may serve as diagnostic and therapeutic targets [88,89,90]. For instance, the chemokine-like factor family with sequence similarity 3, member D (FAM3D), a dual FPR1/FPR2 agonist, is markedly upregulated in human AAA tissues [90], and cinnamoyl-F-(D)L-F-(D)L-F-K (cFLFLF), a PEGylated peptide ligand that binds FPR1 on activated neutrophils, allows the early, accurate and noninvasive diagnosis of AAA [91].
Platelets are the main cells which regulate hemostasis and thrombosis. They play crucial roles in mediating MI, stroke, and venous thromboembolism (VTE). Platelets are also involved in the “immunothrombosis”, a coordinated intravascular coagulation response in which platelets and immune cells prevent dissemination of pathogens leading to activation of the innate and adaptive immune response [92]. Consistently, several viral or bacterial infections contribute to the risk of thrombosis that manifests as arterial thrombosis or VTE. Therefore, although immunothrombosis is an efficient way of assisting the immune system, it may significantly contribute to the overall risk of cardiovascular disease [92]. FPR1 is also expressed on the platelets membrane, and the binding of N-formyl peptides triggers their chemotactic and migratory response [93]. For instance, N-fMLF primes platelet activation increasing the risk of thrombus formation. Fpr1-deficiency in mice or inhibition using a pharmacological inhibitor in human platelets results in diminished agonist-induced platelet activation [94]. This suggests that FPR1 influences the modulation of platelet activation and thrombus formation and could represent a new molecular target able to control platelet-mediated complications in several pathological settings.
ANXA1, that binds both FPR1 and FPR2, plays a modulatory role in platelets. In fact, ANXA1 administration significantly decreases both platelet adherence to the inflamed cerebral endothelium after stroke and regulates the state of platelet activation. ANXA1 also reduces platelet–platelet aggregate formation, decreases thromboxane B2 production and phosphatidylserine expression and, thus, the prothrombotic potential of platelets. It also reduces the propensity for platelets to aggregate and causes thrombosis by affecting integrin αIIbβ3 levels, thereby reducing the risk of thrombotic events. These observations show a multifaceted role for ANXA1 to act both as a therapeutic and a prophylactic drug via its ability to promote endogenous proresolving, anti-thromboinflammatory circuits in cerebral IRI [95].
Table 1 summarizes the role of FPR1 and Figure 1 shows the intracellular signaling pathways mainly involved in cardiovascular diseases (CVDs).
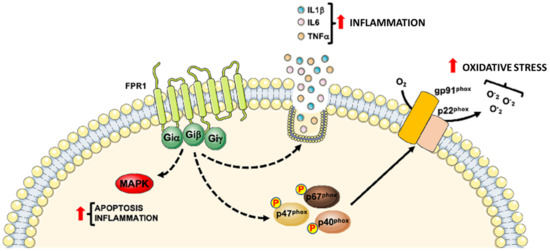
Figure 1. Detrimental role of Formyl Peptide Receptor 1 (FPR1) in CVDs progression. FPR1 contributes to CVDs progression by promoting the inflammatory state through the activation of MAPKs pathway, generation of reactive oxygen species and the release of pro-inflammatory cytokines.
Table 1. FPR1 involvement in cardiovascular diseases (CVDs).
| CVDs | Expression and/or Stimulation | Role |
|---|---|---|
| Ischemia reperfusion injury (IRI) | FPR1 deficiency m | Protective effects by reducing the risk of heart injury induced by IRI [65] |
| FPR1 silencing r | Protective effects mediated by depression of inflammation, cardiomyocyte apoptosis and ventricular remodeling in rats with I/R injury through the suppression of the MAPK signaling pathway activation [66] | |
| FPR1 antagonist CSH m | Protective effects by reducing hepatocyte necrosis/apoptosis, and diminishing inflammatory cytokine, chemokine, and oxidative stress levels as well as accumulation of neutrophils in the necrotic area [67] | |
| FPR1 stimulation with ANXA1 m and r | Cardioprotective role by preserving inotropic responsiveness at the level of ventricular muscle and contractile function of cardiac muscle in vitro [70,71,72,73] | |
| FPR1 blockade m | Beneficial effects mediated by abrogation of reperfusion-induced exacerbation of infarct size [78] | |
| Blood pressure (BP) levels | FPR1 C32T (rs5030878) single nuclear polymorphism (SNP) h | Negative prognostic factor and detrimental effects associated with increased C-reactive protein (CRP) levels linearly related with BP [83,84,85] |
| Abdominal aortic aneurysm (AAA) | FPR1 involvement m | Detrimental effects since FPR1 results involved in neutrophil recruitment and aggravated AAA development [91] |
| Acute myocardial infarction (AMI) | FPR1 as a differentially expressed gene (DEG) h | Role has to be determined even if FPR1 results a DEG in human AMI blood tissue, compared with normal blood tissue using microarray data [75] |
| Potential beneficial role for AMI prevention [76] | ||
| Platelet-mediated complications | FPR1 inhibition or gene deletion m and h | Detrimental effects by impairing agonist-induced platelet activation in Fpr1-deficient mice or in pharmacologically FPR1 inhibited human platelets [94] |
| Endothelial cell function and HUVECs | FPR1 stimulation with NfMLF hc | Beneficial effects by promoting proliferation and capillary network formation |
| Angiogenesis | FPR1 stimulation with ANXA1 hc | Beneficial effects by inducing angiogenesis and/or production of angiogenic factors [56,57,58] |
Table legend: m = mouse; r = rat; h = human; hc = human cell.
This entry is adapted from the peer-reviewed paper 10.3390/life11030243