Human Immunodeficiency Virus type 1 (HIV-1) is a lentivirus containing two RNA genomes that must be reverse transcribed into double-stranded DNA and then integrated into the host genome to ensure a productive infection. HIV-1 can infect host CD4+ T cells and macrophages, resulting in CD4+ T cell loss and immune dysfunction that leads to the acquired immune deficiency syndrome (AIDS).
- HIV-1 capsid
- host factors
- capsid-targeting inhibitors
1. Introduction
HIV-1 has evolved to usurp the host machinery to replicate and spread, as well as to counteract host immune defenses. These processes are achieved mainly from the physical interactions between viral and host cellular proteins, which contribute to the uniqueness of HIV-1 infection [1][2].
HIV-1 infection relies on the viral envelope to bind to the host CD4 receptor and CXCR4 or CCR5 coreceptors for attachment and fusion, resulting in the release of the viral core, capsid, into the cellular cytoplasm [3]. Capsid is comprised of ~250 hexamers and exactly 12 pentamers, assembled from ~1500 copies of monomeric capsid proteins (monomer is defined as CA herein) [4]. This conical capsid houses the viral RNA genome, and the replicative enzymes reverse transcriptase (RT) and integrase (IN), which are required for reverse transcription and integration [5]. Once capsid is present in the cytoplasm, it traffics along the microtubule network to the nuclear envelope, and then is imported into the nucleus in an intact or nearly intact state via the nuclear pore complex (NPC) [6][7]. Reverse transcription appears to be completed in the nucleus, followed by complete uncoating of the capsid and integration of the viral DNA into the host cell genome [8]. However, the exact mechanisms of capsid nuclear entry and when uncoating starts remain to be determined. These events are the early stages of the HIV-1 replication cycle (see Figure 1 for an overview). The successfully integrated viral genome is then transcribed and translated into viral proteins by host machinery to generate new infectious virions, defined as late stages of the HIV-1 replication cycle [9].
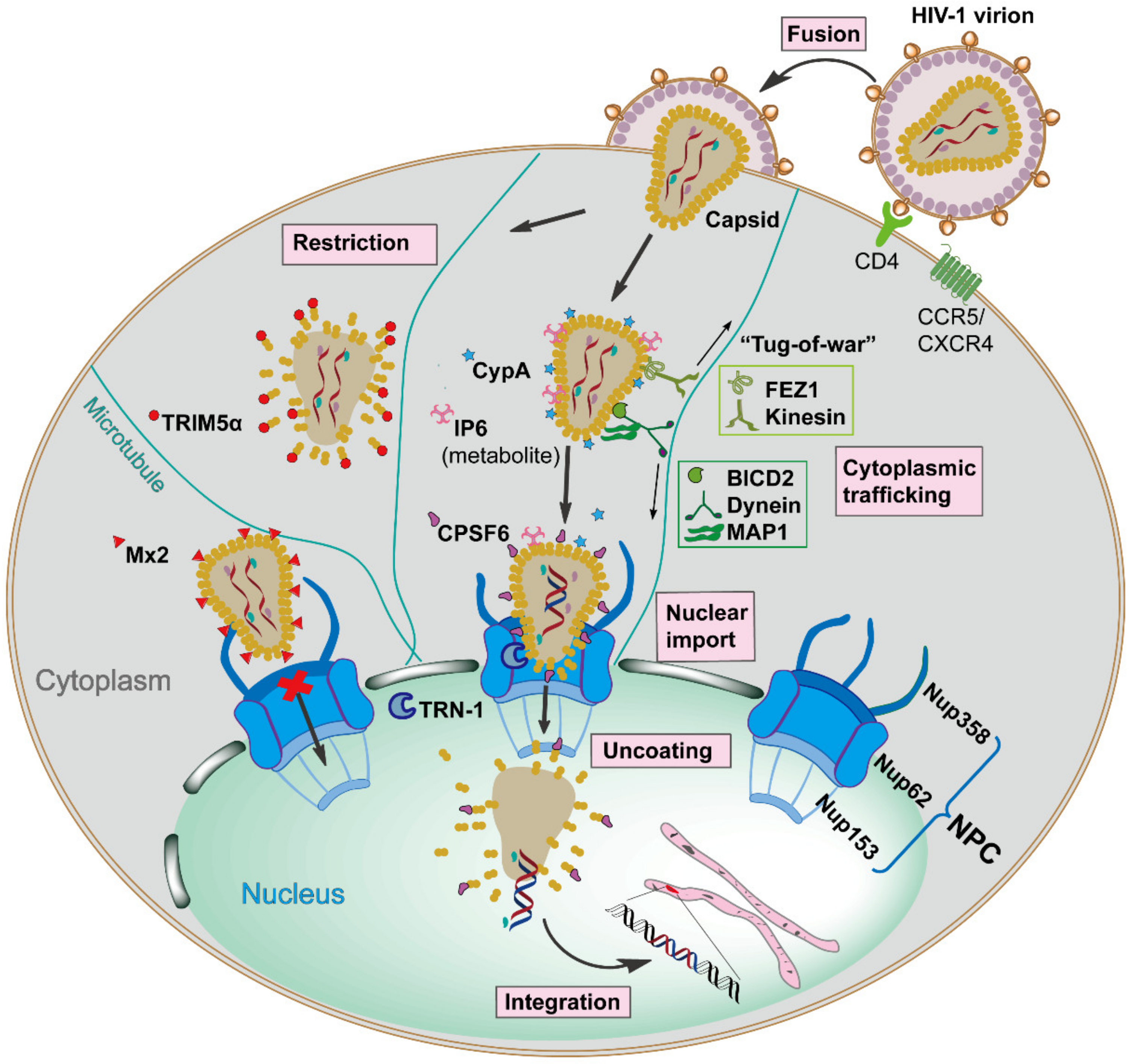
Capsid performs a protective role by shielding the viral genome from host innate immune detection and provides a micro-environment for reverse transcription [10][11]. Moreover, the exterior surface of the capsid has functions beyond the simple encapsidation by providing an expansive area for recognition by diverse cellular proteins acting as either pro-viral or anti-viral factors [12][13][14][15]. Noticeably, these host factors specifically bind to unique high-order interfaces only present in the assembled capsid lattice, and they have no or low affinity to CA monomers, underscoring the fact that capsid structural integrity is critical for the interplay with host factors [16].
Antiretroviral therapy (ART) has turned AIDS from a ‘virtual death sentence’ to a ‘chronic manageable disease’ by targeting and inhibiting the function of viral enzymes (reverse transcriptase, integrase, and protease) [17]. However, the emergence of ART resistance, the absence of an effective vaccine, and the need for long-lasting anti-retroviral compounds to enhance treatment adherence, are reasons for continued research to develop new therapeutics [18][19][20]. A number of non-viral enzymatic targets have been investigated as possible antiretroviral targets. One successful example is the development of the co-receptor antagonist maraviroc which blocks the cellular entry of HIV-1 by preventing the interaction between viral envelope and CCR5 [21][22]. This is the first and only Food and Drug Administration (FDA)-approved anti-HIV drug targeting a host cellular protein. Due to the multiple roles capsid plays during infection, it has been exploited as a target for anti-retroviral compound screening [23][24]. A handful of compounds have been reported to bind capsid and disrupt infection, of which, PF-3450074 (PF74) and the Gilead compounds, GS-CA1 and GS-6207, are potent inhibitors [25]. GS-6207 has entered clinical trials [26]. These potent capsid inhibitors have similar scaffolds and share the same capsid binding pocket with the host proteins CPSF6 and Nup153, which both mediate capsid nuclear entry [26]. These findings underscore the value of investigating whether additional host factor-binding sites on capsid can be identified and exploited for novel therapeutic interventions.
Further to the point of identifying and investigating capsid–host factor binding, which may allow small molecule targeting, HIV–host factor identification studies have benefited significantly from the application of high-throughput “-omic” screening techniques [27][28].
2. HIV-1 Capsid Architecture
The viral capsid shell consists of roughly 1200–1500 monomeric capsid proteins (named CA) assembled into approximately 250 CA hexamers and exactly 12 CA pentamers producing fullerene cone geometry [4][29][30] (Figure 2A–D). Five and seven CA pentamers incorporated at the narrow and broad end of the cone respectively, close the capsid and induce the characteristic curvature. This cone-like capsid measures ~60 nm at its broad end and tapers to 20–30 nm (Figure 2D). However, capsid shapes can vary from the cone-like geometry to tube-like structures with capped ends [31]. As an α-helical protein, each CA has two structurally distinct domains, an N-terminal domain (CA-NTD) and a C-terminal domain (CA-CTD), connected by a flexible linker (Figure 2A). The CA-NTD contains seven α-helices (α1–α7), a characteristic extended cyclophilin A (CypA)-binding loop and a β-hairpin, whereas CA-CTD consists of four α-helices (α8–α11), a short 310-helix, and the major homology region (MHR). The assembly and stability of the capsid shell is driven by three sets of intermolecular protein–protein interactions between CA subunits: (1) intra-hexameric NTD-NTD contacts between individual CA molecules stabilize the hexamers and pentamers that function as the building blocks of the capsid, as well as form a central pore gated by the β-hairpin, (2) intra-hexameric NTD-CTD contacts between adjacent CA molecules further stabilize the individual hexamers and pentamers, and (3) inter-hexameric CTD-CTD contacts participate in dimeric and trimeric interactions to link the individual hexamers and pentamers [31][32][33] (Figure 2D). Therefore, the capsid core not only shields the viral genome but also provides expansive and diverse interfaces for cellular protein interactions. The unique capsid architecture underlies intrinsic core stability and provides for capsid–host factor interactions.
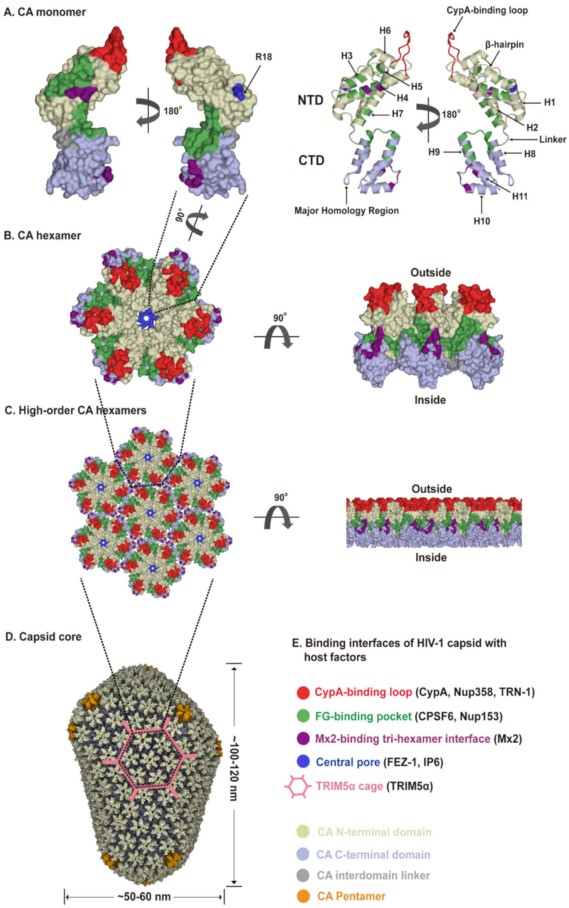
Figure 2. HIV-1 capsid architecture and summary of binding interfaces of capsid with diverse host factors. During the maturation of HIV-1 virion, ~1500 CA monomers assemble into ~250 hexamers and exactly 12 pentamers in alignment produce fullerene cone geometry, forming capsid (Figure 2A–D). The interfaces within the capsid which bind diverse host factors are highlighted in different colors, as detailed in the Figure 2E legend. (A) Structure of the CA monomer (PDB ID: 4XZF). The left structure is shown in surface representation and the right structure as ribbon representation. CA consists of two α-helical domains, an N-terminal domain (NTD) labeled in gold and a C-terminal domain (CTD) labeled in light blue, connected by a flexible linker (grey). All α-helices (H1-H11) and key structural elements are indicated by arrows with names. (B) Structure of a CA hexamer with top view (left) and side view (right) (PDB ID: 4XZF). (C). Structure of high-order (hexagonal) CA hexamers with top view (left) and side view (right) (PDB ID: 4XZF). (D). Architecture of the capsid (PDB ID: 3J3Y). The incorporation of pentamers (orange) at either end of capsid provides the curvature necessary to close the conical structure. (E) The interfaces within capsid that can bind diverse host factors are highlighted in different colors, which are shown in Figure 2A–D. Host proteins CypA, Nup358, and TRN-1 bind to the flexible CypA-binding loop (labeled in red) exposed on the top of CA-NTD that protrudes from the capsid outer surface. Host proteins CPSF6 and Nup153 share the same phenylalanine-glycine binding pocket (labeled in green) created by the NTD-CTD interface between two neighboring CA monomers in a hexamer. The restriction factor Mx2 specifically binds to the three-fold inter-hexamer interface (labeled in purple) of capsid; moreover, it is unable to bind to a CA monomer or a single hexamer. Host protein FEZ1 and metabolite IP6 can bind to the positively charged central pore of hexamers formed by R18 (labeled in dark blue) of CA. TRIM5α can form a hexagonal network (labeled in pink, shown in Figure 2D, also called TRIM5α cage) that avidly binds the capsid shell. The extent to which the TRIM5α cage can cover the capsid and how TRIM5α directly contacts the capsid surface have not been fully established.
This entry is adapted from the peer-reviewed paper 10.3390/v13030417
References
- Friedrich, B.M.; Dziuba, N.; Li, G.; Endsley, M.A.; Murray, J.L.; Ferguson, M.R. Host factors mediating HIV-1 replication. Virus Res. 2011, 161, 101–114.
- Ramdas, P.; Sahu, A.K.; Mishra, T.; Bhardwaj, V.; Chande, A. From entry to egress: Strategic exploitation of the cellular processes by HIV-1. Front. Microbiol. 2020, 11.
- Chen, B. Molecular mechanism of HIV-1 entry. Trends Microbiol. 2019, 27, 878–891.
- Zhao, G.; Perilla, J.R.; Yufenyuy, E.L.; Meng, X.; Chen, B.; Ning, J.; Ahn, J.; Gronenborn, A.M.; Schulten, K.; Aiken, C. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 2013, 497, 643–646.
- Hehl, E.A.; Joshi, P.; Kalpana, G.V.; Prasad, V.R. Interaction between human immunodeficiency virus type 1 reverse transcriptase and integrase proteins. J. Virol. 2004, 78, 5056–5067.
- Gaudin, R.; de Alencar, B.C.; Arhel, N.; Benaroch, P. HIV trafficking in host cells: Motors wanted! Trends Cell Biol. 2013, 23, 652–662.
- Zila, V.; Margiotta, E.; Turonova, B.; Müller, T.G.; Zimmerli, C.E.; Mattei, S.; Allegretti, M.; Börner, K.; Rada, J.; Müller, B. Cone-shaped HIV-1 capsids are transported through intact nuclear pores. bioRxiv 2020.
- Dharan, A.; Bachmann, N.; Talley, S.; Zwikelmaier, V.; Campbell, E.M. Nuclear pore blockade reveals that HIV-1 completes reverse transcription and uncoating in the nucleus. Nat. Microbiol. 2020, 1–8.
- Sundquist, W.I.; Kräusslich, H.-G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924.
- Altfeld, M.; Gale, M., Jr. Innate immunity against HIV-1 infection. Nat. Immunol. 2015, 16, 554.
- Yin, X.; Langer, S.; Zhang, Z.; Herbert, K.M.; Yoh, S.; König, R.; Chanda, S.K. Sensor Sensibility—HIV-1 and the Innate Immune Response. Cells 2020, 9, 254.
- Campbell, E.M.; Hope, T.J. HIV-1 capsid: The multifaceted key player in HIV-1 infection. Nat. Rev. Microbiol. 2015, 13, 471–483.
- Yamashita, M.; Engelman, A.N. Capsid-Dependent host factors in HIV-1 infection. Trends Microbiol. 2017, 25, 741–755.
- Novikova, M.; Zhang, Y.; Freed, E.O.; Peng, K. Multiple roles of HIV-1 capsid during the virus replication cycle. Virol. Sin. 2019, 34, 119–134.
- Temple, J.; Tripler, T.N.; Shen, Q.; Xiong, Y. A snapshot of HIV-1 capsid-host interactions. Curr. Res. Struct. Biol. 2020, 2.
- Summers, B.J.; Digianantonio, K.M.; Smaga, S.S.; Huang, P.-T.; Zhou, K.; Gerber, E.E.; Wang, W.; Xiong, Y. Modular HIV-1 capsid assemblies reveal diverse host-capsid recognition mechanisms. Cell Host Microbe 2019, 26, 203–216.e6.
- Broder, S. The development of antiretroviral therapy and its impact on the HIV-1/AIDS pandemic. Antivir. Res. 2010, 85, 1–18.
- Lu, D.; Lu, T. High Active Anti-retroviral Therapy for HIV/AIDS, Progresses and Drawback. Adv. Pharmacoepidemiol. Drug Saf. 2012, 1, e115.
- Arts, E.J.; Hazuda, D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161.
- Trovato, M.; D’Apice, L.; Prisco, A.; De Berardinis, P. HIV vaccination: A roadmap among advancements and concerns. Int. J. Mol. Sci. 2018, 19, 1241.
- Fätkenheuer, G.; Pozniak, A.L.; Johnson, M.A.; Plettenberg, A.; Staszewski, S.; Hoepelman, A.I.; Saag, M.S.; Goebel, F.D.; Rockstroh, J.K.; Dezube, B.J. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat. Med. 2005, 11, 1170–1172.
- Dorr, P.; Westby, M.; Dobbs, S.; Griffin, P.; Irvine, B.; Macartney, M.; Mori, J.; Rickett, G.; Smith-Burchnell, C.; Napier, C. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 2005, 49, 4721–4732.
- Carnes, S.K.; Sheehan, J.H.; Aiken, C. Inhibitors of the HIV-1 capsid, a target of opportunity. Curr. Opin. HIV AIDS 2018, 13, 359.
- Dick, A.; Cocklin, S. Recent Advances in HIV-1 Gag Inhibitor Design and Development. Molecules 2020, 25, 1687.
- McArthur, C.; Gallazzi, F.; Quinn, T.P.; Singh, K. HIV Capsid Inhibitors Beyond PF74. Diseases 2019, 7, 56.
- Bester, S.M.; Wei, G.; Zhao, H.; Adu-Ampratwum, D.; Iqbal, N.; Courouble, V.V.; Francis, A.C.; Annamalai, A.S.; Singh, P.K.; Shkriabai, N. Structural and mechanistic bases for a potent HIV-1 capsid inhibitor. Science 2020, 370, 360–364.
- Jaeger, S.; Cimermancic, P.; Gulbahce, N.; Johnson, J.R.; McGovern, K.E.; Clarke, S.C.; Shales, M.; Mercenne, G.; Pache, L.; Li, K. Global landscape of HIV–human protein complexes. Nature 2012, 481, 365–370.
- Ivanov, S.; Lagunin, A.; Filimonov, D.; Tarasova, O. Network-Based Analysis of OMICs Data to Understand the HIV-Host Interaction. Front. Microbiol. 2020.
- Ganser-Pornillos, B.K.; Yeager, M.; Sundquist, W.I. The structural biology of HIV assembly. Curr. Opin. Struct. Biol. 2008, 18, 203–217.
- Pornillos, O.; Ganser-Pornillos, B.K.; Kelly, B.N.; Hua, Y.; Whitby, F.G.; Stout, C.D.; Sundquist, W.I.; Hill, C.P.; Yeager, M. X-ray structures of the hexameric building block of the HIV capsid. Cell 2009, 137, 1282–1292.
- Mattei, S.; Glass, B.; Hagen, W.J.; Kräusslich, H.-G.; Briggs, J.A. The structure and flexibility of conical HIV-1 capsids determined within intact virions. Science 2016, 354, 1434–1437.
- Gres, A.T.; Kirby, K.A.; KewalRamani, V.N.; Tanner, J.J.; Pornillos, O.; Sarafianos, S.G. X-ray crystal structures of native HIV-1 capsid protein reveal conformational variability. Science 2015, 349, 99–103.
- Kleinpeter, A.B.; Freed, E.O. HIV-1 maturation: Lessons learned from inhibitors. Viruses 2020, 12, 940.