T-lymphokine-activated killer cell-originated protein kinase (TOPK), also known as PDZ-binding kinase (PBK), was a member of the MEK3/6-related MAPKK family. As a mitotic serine/threonine protein kinase, accumulating evidence supported its role in mitosis and cell-cycle progression of mitotically active cells, especially proliferative malignant cells. PBK/TOPK was confirmed to be associated with the development, progression, and metastasis of malignancies, which made it a potential therapeutic target in cancer therapy. Further, it was also demonstrated to play crucial roles in ischemic injury and involve in protection against ischemia. This protective effect of PBK/TOPK in the context of ischemia challeged the development of PBK/TOPK inhibitors in anti-tumor therapy, and more research was required to further explore its role and underlying mechanisms to translate its application to clinical studies.
- PBK/TOPK
- mitosis
- malignancy
- ischemia
- inhibitor
1. Introduction
T-lymphokine-activated killer-cell-originated protein kinase (TOPK), also known as PDZ-binding kinase (PBK), is a novel mitotic serine/threonine protein kinase [1,2]. PBK/TOPK is overexpressed in various actively proliferative cells, including malignant tumor cells, as well as normal cells, such as sperm cells. The transcription, activation, and degradation of PBK/TOPK is regulated by mitosis progression, which is mediated by a series of proteins. As a mitotic kinase, PBK/TOPK is important for the proliferation, progression, and metastasis of many cancers, including leukemia and myeloma, among others [3,4]. Upregulation of PBK/TOPK has also been proven to be associated with cancer diagnosis and prognosis, and, thus, it may be a potential therapeutic target in various malignant tumors. Thus, various potent PBK/TOPK inhibitors have been developed. The function of PBK/TOPK as an emerging target for cancer-specific therapeutics has been reviewed previously by Herbert et al. [5]. Furthermore, ours and other research teams also demonstrated that PBK/TOPK plays crucial roles in ischemic injury and is involved in protection against ischemia and ischemic postconditioning [6,7,8]. This protective effect of PBK/TOPK in the context of ischemia challenged the development of PBK/TOPK inhibitors in antitumor therapy, and more research is required to further explore its role and underlying mechanisms, to translate its applicability to clinical studies.
2. Identification of PBK/TOPK
The human homologue of the Drosophila tumor suppressor Discs-large (hDlg) is a membrane-associated guanylate kinase homologue (MAGUK) [9]. Several proteins that bind to hDlg have been demonstrated to be involved in cell growth control, including in the pathogenesis of adenomatous polyposis coli [10,11,12,13]. In 2000, Gaudet et al. sought to elucidate the signal transduction pathway through which hDlg regulates cellular proliferation, and they first identified a 322 amino acid serine/threonine kinase, using a two-hybrid screen and named it PDZ-binding kinase (PBK) [2]. At the same time, Abe et al. cloned a novel protein kinase from a lymphokine-activated killer T (T-LAK) cell subtraction cDNA fragment library and named it T-LAK cell-originated protein kinase (TOPK) [1]. This gene was later confirmed to be similar to that found by Gaudet et al., and this novel gene, or its protein product, was called PBK/TOPK.
3. General Features of PBK/TOPK
Unlike hDlg, which is ubiquitously expressed, the expression and distribution of PBK/TOPK varies. It is abundant in highly proliferative tissues, such as the placenta, testis, T-LAK cells, activated lymphoid cells and lymphoid tumors, but it is expressed at a very low level or is even absent in non-proliferative normal tissues, such as normal adult brain tissue (with the exception of the subependymal zone and early postnatal cerebellar external layer, which is enriched with rapidly proliferating progenitor cells) [14,15]. Regarding its intracellular distribution, PBK/TOPK is expressed both in the cytoplasm and the nucleus; however, it may be expressed exclusively in the nucleus of tumor cells or in mitosis, particularly around chromosomal surfaces during prophase and metaphase [16,17]. Moreover, both PBK/TOPK protein expression and its activation are subject to cell-cycle regulation. Using the thymidine double-block method, PBK/TOPK protein expression was demonstrated to increase and peak at 8 h after being released from the cell-cycle block during G2 to M phase in HeLa cells, and this was accompanied by the upregulation of its phosphorylation activity [18].
As a member of the MEK3/6-related MAPKK family, PBK/TOPK shares the characteristic serine/threonine protein kinase subdomains and a C-terminal PDZ-binding T/SXV motif, which enables it to bind specifically to the PDZ2 domain of hDlg or other PDZ-containing proteins. Activated PBK/TOPK is also able to phosphorylate P38 MAPK and histone H3, among other proteins [2,19,20]. However, the phosphotransferase activity of PBK/TOPK appears to be regulated in a cell-cycle-dependent manner; that is, it is activated following its phosphorylation by cyclin-dependent kinase 1 (CDK1)/cyclin B1 exclusively at mitosis (Figure 1). Notably, this process depends on the binding of PBK/TOPK to the CDK1/cyclin B1 complex at the mitotic spindle, which in turn induces its phosphorylation and thereby increases its binding ability to the CDK1/cyclin B1 complex through phosphorylation and inactivation of protein phosphatase 1 alpha (PP1α) [2,21,22]. These results all suggest that PBK/TOPK may be involved in the regulation of cellular proliferation and cell-cycle progression.
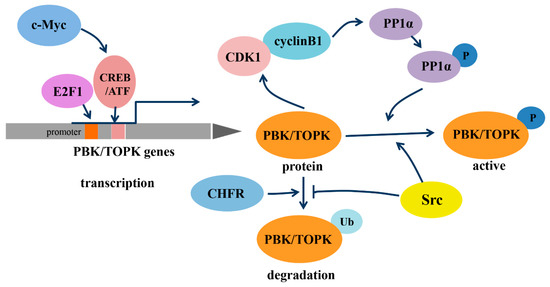
In addition, the transcription of PBK/TOPK and its degradation varies with cell-cycle progression (Figure 1). The binding of the cell-cycle-specific transcription factors E2F and cyclic AMP-responsive element-binding protein/activating transcription factor (CREB/ATF) to the −146 and −312 bp binding sites within the PBK/TOPK promoter directly leads to its transcriptional upregulation [17,23]. C-Myc is functionally linked with E2F1 in controlling cell-cycle progression, and it activates PBK/TOPK transcription by indirectly enhancing E2F1 activity and cooperatively binding with E2F1 [24]. Checkpoint protein with FHA and RING domains (CHFR) is an E3 ubiquitin ligase that was demonstrated to ubiquitinate and mediate PBK/TOPK degradation, which is essential for its mitotic checkpoint function [25]. Src, a transforming protein with tyrosine kinase activity, was also identified as an upstream regulator of PBK/TOPK. It can directly bind and phosphorylate PBK/TOPK at Y74 and Y272, thereby enhancing its activity and enhancing PBK/TOPK stability, which allows it to avoid degradation after ubiquitination [26].
4. PBK/TOPK Function in Mitotic Progression and Tumor Cellular Proliferation
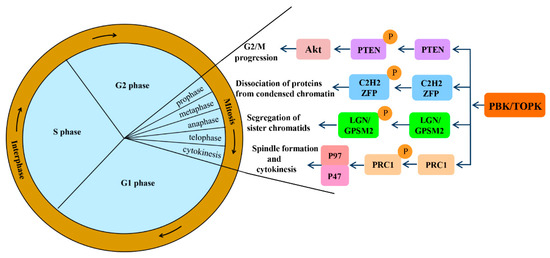
Mitotic progression is regulated by the co-ordination of several proteins and is crucial for the maintenance of genomic stability. PBK/TOPK is known to play pivotal roles in cellular mitosis; its main mitotic substrates and detailed functions in mitosis have been studied in depth (Figure 2). PBK/TOPK is of crucial importance in CHFR-mediated G2/M progression via phosphorylation and inactivation of PTEN, which results in activation of Akt [25]. Activated TOPK was also confirmed as the principal C2H2 zinc finger protein (ZFP) linker kinase that phosphorylates C2H2 ZFPs linkers within minutes in the prophase stage. This is a unique mechanism for reducing the DNA-binding activity of C2H2 ZFPs, which mediates the dissociation of hundreds of proteins from condensed chromatin during transition from prophase to telophase. PBK/TOPK was the first kinase to be identified as a master regulator of an entire family of transcription factors, based on their conserved motif [27]. At the late stage of mitosis, upregulated PBK/TOPK was shown to mediate the phosphorylation of LGN/GPSM2 (Leu-Gly-Asn repeat-enriched protein/G-protein signaling modulator 2) at Thr450, which might play a critical role in the segregation of sister chromatids and the formation of F-actin polymerization at the contractile ring [28]. In addition, activated PBK/TOPK was demonstrated to enhance the phosphorylation of the microtubule-binding protein PRC1 and ATPase protein p97 with p47 as an adaptor protein, which is indispensable for mitotic spindle formation and cytokinesis during mitosis [29].
PBK/TOPK overexpression in oncogenic pathways has been implicated in the proliferation, metastasis, and anti-apoptosis of tumors (Figure 3). Its overexpression allows the tumor cells to bypass the natural surveillance mechanism associated with the G2/M checkpoint and leads to aberrant entry into the mitotic phase by phosphorylating histone H3 at Ser10, as well as down-modulating the tumor suppressor p53, upregulating the cyclin-dependent kinase inhibitor p21, and thereby contributing to tumorigenesis [30,31,32]. Conversely, suppression of PBK/TOPK impaired its tumorigenic properties by reducing the phosphorylation and activation of mitogen-activated protein (MAP) kinase, including extracellular signal-regulated kinase (ERK) 2 and P38, inhibiting the expression of mutant p53, and inhibiting Akt activation [33,34,35,36,37,38]. Notably, phosphorylated ERK2 could in turn phosphorylate PBK/TOPK and increase its kinase activity, resulting in a positive feedback loop between PBK/TOPK and ERK2. PBK/TOPK also possesses the capacity to activate the interaction of transcriptional factor β-catenin, with its transcriptional coactivators T-cell factor/lymphoid enhancer-binding factor (TCF/LEF), which subsequently upregulates the transcription of matrix metalloproteinase MMP-2 and MMP-9, thereby facilitating the invasiveness and metastasis of tumor cells [16].

Aside from promoting proliferation and metastasis, activated PBK/TOPK could also alleviate As3+ treatment–induced apoptosis by binding with and phosphorylating histone H2AX at Ser139 in the nucleus. This may be responsible for the resistance of some melanoma cells to As3+ treatment [39]. Moreover, PBK/TOPK could inhibit UVB-induced apoptosis as well, by phosphorylating c-Jun-NH2-Kinase 1 (JNK1) at Thr183/Tyr185, activating it, and increasing the peroxidase activity of peroxiredoxin1 (Prx1) and decreasing the intracellular accumulation of H2O2 via phosphorylation Prx1 at Ser32 [40,41]. Moreover, TOPK directly phosphorylates inhibitor-κBα (IκBα) at Ser 32, which induces P65 nuclear translocation, nuclear factor-kappa B (NF-κB) activation, and subsequently leads to responsive transcription of anti-apoptotic genes [42,43,44]. These findings enrich our knowledge of the regulatory functions and underlying mechanisms of PBK/TOPK in mitotic progression of actively proliferating cells, especially tumors, and have led to a better understanding of its role in tumorigenesis. These studies all suggest that PBK/TOPK might serve as a diagnostic/prognostic indicator and therapeutic target in tumors.
This entry is adapted from the peer-reviewed paper 10.3390/cells10020371