The phosphoinositide 3-kinase (PI3K) is a family of kinases that play a key role in the biology of chronic lymphocytic leukemia (CLL).
- PI3K inhibitors
- chronic lymphocytic leukemia
- targeted therapy
1. Introduction
Chronic lymphocytic leukemia (CLL) is a slow-growing cancer that primarily afflicts elderly population and represents the most common type of leukemia in Western countries [1]. CLL induces B-cell expansion in blood, bone marrow and secondary lymphoid tissues that is primarily driven by B-cell receptor (BCR) signaling.
The phosphoinositide 3-kinase (PI3K) family plays a key node in BCR pathway promoting proliferation and survival in B cell malignancies, including CLL [2]. Advances in our understanding of the role of PI3K in CLL led to the development of modern pathway inhibitors which are revolutionizing the treatment landscape of this disease. The main targets for PI3K inhibition are the four isoforms α, β, γ, and δ, collectively regulating CLL cell chemotaxis, cytoskeletal rearrangement and the interaction of leukemic cells with the tumor microenvironment.
The potential efficacy of small molecule PI3K inhibitors (PI3Kis) has been widely recognized in CLL therapy [3]. However, several issues have become pertinent with the availability of PI3K inhibitors and the limits arising are of similar magnitude as the advances that have been made. In particular, toxic side effects can significantly impair patients’ quality of life as well as treatment efficacy.
2. The PI3K in CLL: BCR-Signaling and Beyond
PI3Ks represent a family of highly conserved lipid kinases which generate second messengers by specific catalytic 3-hydroxy phosphorylation of phosphatidylinositol (PI) [4]. PI3Ks are divided into four classes based on their in vitro lipid substrate specificity and structure [5]. Class I is the subfamily most implicated in human cancer and functions as a dimeric enzyme consisting of catalytic and regulatory subunits. Mammals express four tissue-specific Class I catalytic isoforms (p110 α, β, γ and δ) that are associated with various tissue pathways. PI3K isoforms p110α and p110β are ubiquitously expressed [6,7,8], while γ and δ isoforms are highly enriched in leukocytes [9,10,11], where they have distinct and nonoverlapping roles in immune cell development and function. Therefore, tissue distribution informs the expected activity and toxicity seen with pharmacologic inhibition of these different isoforms.
PI3Ks are a part of the intracellular PI3K/AKT/mammalian target of the rapamycin (mTOR) signaling axis that plays a key role in signal transduction, cell metabolism and survival [8,12]. Importantly, PI3K lies downstream of the B-cell antigen receptor signaling in both normal and malignant B cells (Figure 1), the latter being described to have a constitutively activated PI3K/AKT/mTOR pathway [13].
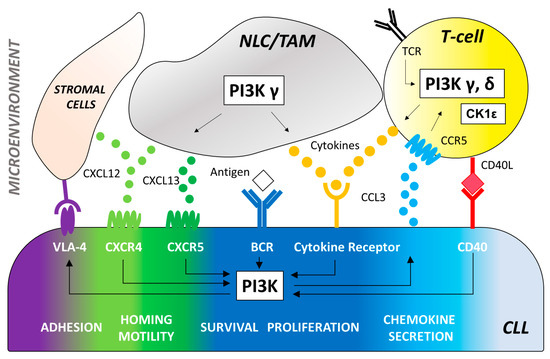
Figure 1. The effector molecule PI3K plays a central role in multiple signaling pathways promoting B-cell development and function. CLL cells depend on signaling via the BCR engagement that induces the activation of PI3K-dependent pathways to increase survival, proliferation, adhesion to stromal cells (via VLA-4), and secretion of chemokines (CCL3 and CCL4). Antigens for the BCR may be expressed by the CLL cells themselves or by other cells in the microenvironment. Key cell types of the tumor microenvironment include: 1. T-cells that can be recruited by CCL3 production and CCR5 receptor binding to provide CD40 ligand (CD40L) and cytokines (IL4, TNFα, IFNγ, CCL3, CCL4) that induce PI3K activation and stimulate the expansion of the malignant CLL clone. PI3K/AKT signaling triggered by the T cell receptor (TCR) plays a critical role in T-cell differentiation, function, and immune tolerance. PI3Kδ controls the activity of regulatory T-cells (Tregs) resulting in enhanced anti-tumoral immune functions which may contribute to the activity and toxicity of PI3Kδ inhibitors in CLL therapy. Impairment of Treg function by PI3Kδ inhibition can be counteracted with CK1ε modulation. 2. Myeloid-derived nurse-like cells (NLC) and tumor-associated macrophages (TAM) that secrete cytokines and chemokines, such as CXCL12 and CXCL13, which bind to CXCR4 and CXCR5 on CLL cells. PI3Kδ is a central integrator of signals from the CXCR4/CXCL12 and CXCR5/CXCL13 axis. 3. Stromal cells of mesenchymal origin that also secrete CXCL12 to facilitate CLL recruitment to and retention in the lymph nodes via VLA-4 binging. *VLA-4 = Very late antigen 4, CCL3 = C-C motif chemokine ligand 3, CCL4 = C-C motif chemokine ligand 4, CCR5 = C-C motif chemokine receptor type 5, CD40L = CD40 ligand, IL4 = Interleukin 4, TNFα = Tumor necrosis factor, INFγ = Interferon gamma, CXCL12 = C-X-C motif chemokine ligand 12, CXCL13 = C-X-C motif chemokine ligand 13, CXCR4 = C-X-C chemokine receptor type 4, CXCR5 = C-X-C chemokine receptor type 5.
CLL cells with unmutated immunoglobulin heavy chain variable region (IGHV) show significantly greater PI3K expression compared to IGHV mutated counterpart [14]. After BCR engagement, the Class I PI3Ks p110γ and p110δ produce the phospholipid phosphatidylinositol-(3,4,5)-trisphosphate (PIP3) [15,16] leading to the activation of several downstream mediators of essential cellular processes, including cell survival, proliferation, and metabolic fitness [11,17]. In B cells, the PH domain of both AKT and BTK allow PIP3 binding to keep an activation loop “open” for substrate docking [18]. Additional downstream effects of PI3K activation include inhibition of IκB Kinase (IKK), Forkhead box O (FOXO), and the proapoptotic protein B-cell lymphoma 2 (BCL2) associated agonist of cell death (BAD) as well as activation of mTOR. The PI3K/AKT signaling can also inhibit the lysosome-mediated degradation of NOTCH1 [19] a new key cancer gene in CLL whose genetic and pathway alterations are likely to represent a novel oncogenic process in this disease [20,21,22,23]. Interestingly, we demonstrated the involvement of the PI3Kδ oncogenic pathway in the phosphorylation of NOTCH1 intracellular domain in CLL [24,25,26]. Besides its role in BCR signaling, PI3Kδ also plays an important role in CLL cell migration and tissue homing. In vitro studies have shown that antigen receptors, costimulatory molecules, cytokine and chemokine receptors signaling can all trigger an increase in PIP3 and phosphorylation of AKT in different B-cell disease [27,28,29] (Figure 1).
The PI3K pathway extends beyond the direct regulation of CLL cell-intrinsic activities through the BCR (Figure 1). It is well known that the tumor biology of CLL cells depends on a complex cross talk with a number of non-neoplastic cells comprising the tumor microenvironment (i.e., mesenchymal stromal cells, nurse-like cells and lymphoma-associated macrophages, in concert with T cells, natural killer cells and extracellular matrix components). Several studies highlighted the relevance of specific PI3K isoform signaling on CLL microenvironment. The PI3K p110δ is critical for survival and function of CD4+CD25+FOXP3+ regulatory T cells (Treg) [30,31]. In mice, PI3Kδ deletion had a significant deleterious effect on the induction of Treg which cannot block the development of experimental colitis [16,32]. Thus, the role of p110δ Treg function may explain some of the toxicities seen with PI3K inhibitors describe thereafter in this review. Interestingly, immune suppression of Tregs can be restored by the inhibition the casein-kinase 1 epsilon (CK1ε), a Wingless-related integration site (WNT) signaling amplifier that negatively modulates regulatory T cell function [33]. This represents an alternative approach to lower the risk of immune-mediated toxicities frequently observed with PI3K p110δ inhibitors. PI3Kγ and δ are essential mediators of chemokine receptor signaling necessary for the interaction of CLL with tissue-resident stromal cells. Specifically, PI3Kγ contributes to the differentiation and migration of key support cells in the tumor microenvironment, such as CD4+ T cells and M2 tumor-associated macrophages, which sustain leukemia cells in a protective niche [34,35]. PI3K p110γ is also critical for many aspects of neutrophil function, including chemotaxis, phagosome formation, and the oxidative burst [36,37]. Additionally, genetic p110δ inhibition enhances toll-like receptor signaling in macrophages [32] and leads to prolonged pro-inflammatory responses in dendritic cells [38]. These data indicate that PI3Ks play a critical role in the innate immune systems as well as the adaptive immune system, suggesting that p110δ and p110γ inhibition may enhance immune responses. PI3K inhibitors efficacy and toxicity is the result of a combination of these pleiotropic effects in the tumor microenvironment with CLL cell-intrinsic activities.
3. Approved PI3K Inhibitors in CLL
3.1. Idelalisib
Idelalsib (CAL-101) is a first-generation inhibitor of the delta isoform of the catalytic subunit of PI3K p110δ, which is preferentially expressed in leukocytes [39] (Figure 2).
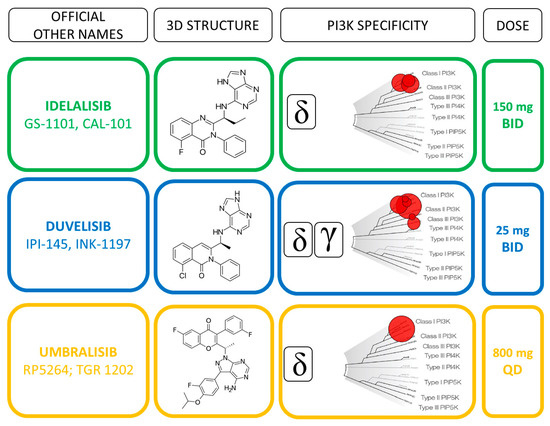
Figure 2. Direct comparison of idelalisib, duvelisib and umbralisib profiles.
Idelalisib inhibited cellular pathways involved in the homing and retention of normal and malignant B cells in lymph nodes and bone marrow, thereby causing transient lymphocytosis and rapid lymph node shrinking [40,41]. The recommended dosage of idelalisib is 150 mg administered orally twice daily. A phase III study evaluated idelalisib and rituximab versus placebo and rituximab therapy in relapsed CLL, with an overall response rate (ORR) of 81% that allowed the first FDA and EMA approval of a PI3K inhibitor in CLL [3]. In a second trial, a population of treatment naïve (TN) elderly patients with TP53 alterations displayed 100% ORR with high PFS and OS rates at 36 months [42]. A recent phase III randomized trial (ASCEND) assessed the efficacy and safety of acalabrutinib compared with idelalisib plus rituximab (idelalisib-R) for R/R CLL [43]. This trial, which is one of the first direct comparison between two inhibitors of the B-cell receptor pathways, demonstrated significantly longer PFS of acalabrutinib monotherapy over idelalisib-R regimen. Acalabrutinib was discontinued in 11% for AEs while idelalisib-R was discontinued in 47% of cases with a median treatment duration of 11.5 months, which was shorter compared with previous studies [3,42]. Interestingly, the ORR was similar between the acalabrutinib and idelalisib-R treatment, confirming that toxicity and early drug discontinuation may have contributed to the improved PFS with acalabrutinib. The authors speculated that a higher rate of discontinuation of idelalisib was correlated either to a better clinical experience which had facilitated an earlier identification of AEs, or to the setting of the patients (i.e., younger and less pretreated with a more intact immune system).
Currently, combination of idelalisib and rituximab is indicated in relapsed/refractory (R/R) CLL and in first-line therapy of patients with del(17p)/TP53 mutations. However, the latter was restricted only to patients not eligible for any other therapies, as alternative treatments have better benefit/risk ratio. Indeed, prolonged follow-up documented serious adverse events during idelalisib administration, including but not limited to a high risk of autoimmune complications (i.e., colitis, pneumonitis and transaminitis) and infections (i.e., cytomegalovirus reactivation and pneumocystis jirovecii pneumonia) [44]. Current limits to the use of idelalisib in clinical practice together with strategies for overcoming these challenges will be discussed in the following paragraphs.
3.2. Duvelisib
Duvelisib (IPI-145) is a dual inhibitor of PI3Kδ and PI3Kγ isoforms (Figure 2) that have been shown to support CLL survival in distinct and independent manners [45]. Preclinical evidence showed that PI3Kδ inhibition directly affects the leukemic B cells, whereas PI3Kγ inhibition targets key support cells in the tumor protective niche, such as CD4+ T cells and M2 tumor-associated macrophages [34,46] (Figure 1). The unique binding affinity to PI3Kγ together with a long target residence time represent distinct features compared to idelalisib that may improve the therapeutic profile of duvelisib [47]. Furthermore, duvelisib overcomes the ibrutinib resistance of treatment-induced BTK C481S mutation in vitro [48].
The recommended oral dose of 25 mg BID of duvelisib was identified in the first phase I study as the most appropriate for patient affected by different hematological malignancies, including treatment naïve (TN; n =18) and R/R (n = 55) CLL [49]. Patients remained on treatment for a median of 62.3 and 24 weeks in TN and R/R cohort, respectively. The achievement of a clinical response was higher in TN (83%) than R/R (56%) patients, while being independent from adverse prognostic features in both cohorts. The efficacy of duvelisib monotherapy was confirmed in the phase III “DUO trial” comparing duvelisib to ofatumumab in 319 R/R CLL, excluding previously BCR inhibitors treated patients from trial [50]. After a median follow-up of 22.4 months, duvelisib resulted superior to ofatumumab in terms of PFS and ORR rates (13.3 vs. 9.9 months and 73.8% vs. 45.3%, respectively). The higher benefit of duvelisib was conserved when considering the subset of patients who received ≥2 prior lines of therapy [51], for which the drug obtained the FDA approval in 2018. Similarly to idelalisib, the safety profile of duvelisib lead to a high rate of treatment discontinuation, thus hampering the therapeutic advantage of this molecule.
(References would be added automatically after the entry is online)
This entry is adapted from the peer-reviewed paper 10.3390/cancers13061280