Protein-bound uremic toxins constitute approximately 25% of all currently identified uremic toxins. As a consequence of being highly bound to plasma proteins, protein-bound uremic toxins are poorly cleared by dialysis.
- uremic toxins
- chronic kidney disease
- cardiovascular disease
- indoxyl sulfate
- p-cresyl sulfate
- hippuric acid
- trimethylamine N-oxide
- asymmetric dimethylarginine
- tumor necrosis factor al-pha
- interleukin 6
1. Introduction
Uremia is a complication of chronic kidney disease (CKD) and is defined as the accumulation of solutes that are normally cleared by the kidneys [1]. If left untreated, uremia is a life-threatening condition. Though dialysis greatly prolongs the survival of end-stage renal disease (ESRD) patients, it is unable to completely mitigate the uremic condition, leaving patients with what is referred to as “residual syndrome” [1]. Despite regular dialysis treatment, the incomplete removal of organic waste compounds results in accumulation of uremic toxins, which play a crucial role in the progression of CKD and cardiovascular disease (CVD). To date, over 100 uremic toxins have been identified and classified by The European Uremic Toxin Work Group (EUTox) [2]. The EUTox classifies uremic toxins by their physicochemical properties into the following categories: free water-soluble low-molecular-weight solutes (<500 Da), protein-bound solutes, and middle molecules (≥500 Da) [2].
2. Protein-Bound Uremic Toxins
Protein-bound uremic toxins constitute approximately 25% of all currently identified uremic toxins [2]. As a consequence of being highly bound to plasma proteins, protein-bound uremic toxins are poorly cleared by dialysis [3]. Many of these protein-bound uremic toxins are gut-derived and are the by-products of aromatic amino acid breakdown by intestinal bacteria.
2.1. Indoxyl Sulfate
Indole is a metabolic product of tryptophan decomposition by bacterial tryptophanase. Once produced by intestinal bacteria, indole is absorbed into the portal circulation and enters the liver (Figure 1) [4]. In the liver, indole is hydroxylated by cytochrome P450 2E1 (CYP2E1) to form 3-hydroxy indole [5] and subsequently sulfated by sulfotransferase 1A1 (SULT1A1) to produce indoxyl sulfate (IS) [6]. IS is extensively excreted in the urine by proximal tubular secretion through basolateral organic anion transporter 1 (OAT1) and OAT3 [7]. IS is highly protein-bound (Table 1) to albumin in the circulation (93%) [8] and is consequently poorly cleared by dialysis [9]. As kidney function declines, IS levels increase in the blood and this elevation contributes to further progression of CKD [10].
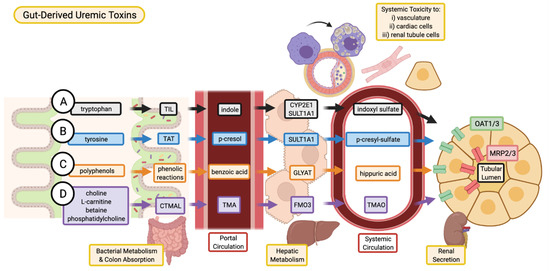
Figure 1. Absorption, and excretion pathway of gut-derived uremic toxins. (A) Indoxyl sulfate is produced when dietary tryptophan is converted into indole by bacterial tryptophan indole-lyase (TIL) and subsequent absorption into the portal circulation for further metabolism by cytochrome P450 2E1 (CYP2E1) and sulfotransferase 1A1 (SULT1A1). (B) P-cresyl sulfate begins as dietary tyrosine which is metabolized by tyrosine aminotransferase (TAT) into p-cresol. These intermediates are converted into p-cresyl sulfate through SULT1A1. (C) Various dietary polyphenols are converted through multiple phenolic reactions by colon bacteria into benzoic acid which is then conjugated with glycine by glycine-N-acyltransferase (GLYAT) to produce hippuric acid. (D) Trimethylamine N-oxide (TMAO) is the product of TMA oxidation by flavin-containing monooxygenase 3 (FMO3), where TMA is the intermediate metabolite of carnitine trimethylamine lyase (CTMAL) breakdown of various dietary molecules such as choline, L-carnitine, betaine, and phosphatidylcholine (TMAO is a non-protein bound, gut-derived uremic toxin and is discussed in detail in the full article: 10.3390/toxins13020142). (A–D) Uremic toxins are secreted by renal tubular cell via drug transporters (organic anion transporter 1/3, OAT1/3; and multidrug resistance-associated proteins 2/3, MRP2/3 and subsequently excreted in the urine. Image created with BioRender.com.
| Molecule | Size (MW) | Protein Binding | Dialyzability | Fold Change (M/N) * | Origin | Site of Toxicity | Mechanism of Toxicity | Therapeutic Interventions |
|---|---|---|---|---|---|---|---|---|
| Indoxyl Sulfate | 213.2 | 93% bound to albumin | 32% cleared through dialysis | 43.2 [2] | Metabolism of tryptophan by colon microbes. | Kidney proximal tubule cells, cardiomyocytes, endothelial cells, and VSMCs. | Generation of ROS, induction of fibrosis/inflammation in kidneys. Induce oxidative stress in VSMC. | AST-120. Pre-, pro-, and synbiotics. Dietary modulation. |
| p-Cresyl Sulfate | 188.2 | 90% bound to albumin | 29% cleared through dialysis | 11.0 [2] | Metabolism of aromatic amino acids by colon microbes. | Kidney proximal tubule cells and endothelial cells. | Generation of ROS, induction of fibrosis/inflammation in kidneys and endothelial cells. | AST-120. Pre-, pro-, and synbiotics. Dietary modulation. |
| Hippuric Acid | 179.2 | 34–40% bound to albumin | 64% cleared through dialysis | 23.8 [2] | Metabolism of dietary polyphenols by colon microbes. | Renal tubular cells and endothelial cells. | Generation of ROS, promotes renal fibrosis and endothelial dysfunction. | Potential interventions: Pre-, pro-, and synbiotics. Dietary modulation. |
| Trimethylamine N-oxide (TMAO) | 75.1 | Free water soluble | 85% cleared though dialysis | 28.6 [11] | Metabolism of dietary precursors choline, phosphatidylcholine, L-carnitine, and betaine by colon microbes. | Renal tubular cells, endothelial cells and VSMCs. | Induction of renal fibrosis. Enhance immune response in atherosclerosis. | Diet modulation. Probiotics. Choline analogues such as DMB, IMC and FMC. |
| Asymmetric Dimethylarginine (ADMA) | 202.3 | 30% bound to albumin [12] | 20–40% cleared through dialysis [13] | >6.4 [2] | Non-proteinogenic amino acid synthesized through post translational methylation of arginine by PRMTs. | Renal tubular cells, vasculature, and cardiomyocytes. | Renal fibrosis, generation of ROSInhibitor of NOS leading to impaired NO signaling. Promote foam cell formation. | Potentiate ADMA metabolism by increasing DDAH activity. Dietary antioxidants (i.e., Quercetin). L-arginine supplementation. |
| Tumor Necrosis Factor alpha (TNF α) | 17,300 | N/A | Minimal | 3.09 [2] | Largely from immune cells (T lymphocytes, macrophages, mast cells), and vascular endothelial cells, renal tubular epithelial and mesangial cells, cardiomyocytes. | Renal proximal tubule, glomerulus and interstitium, vasculature. | Fibrosis, glomerulosclerosis, superoxide generation, macrophage infiltration, vascular calcification, atherosclerosis. | TNF- α blockers (e.g., Adalimumab, etanercept, infliximab). ACE inhibitors (e.g., Captopril). |
| Interleukin- 6 (IL- 6) | 21,000 | N/A | Minimal | 1.48 [2] | Hepatocytes, megakaryocytes, immune cells (neutrophils, B- and some T-cells, monocytes/macrophages). | Renal tubules, glomerulus, interstitium, cardiac fibroblasts and myocytes, vasculature | Renal fibrosis, cardiac fibrosis (left ventricular hypertrophy), atherosclerosis. | Neutralization of soluble and membrane bound IL-6 receptors (e.g., Tocilizumab) and gp130 (e.g., Bazedoxifene). |
2.1.1. Mechanisms for the Progression of CKD
IS has shown nephrotoxic effects through generation of reactive oxygen species (ROS) [14], depletion of anti-oxidative systems [15], and induction of fibrosis and inflammation (Figure 2). NF-κB plays a central role in the pathological effects of IS in the kidneys. In human proximal tubule cells (HK-2) the activation of NF-κB by IS suppressed cellular proliferation, induced and accelerated senescence through induction of p53, and promoted fibrosis by inducing TGF-β1 and PAI-1 expression [14][16]. p53 induction was also proposed to contribute to renal fibrosis by stimulating the expression of TGF-β1 and subsequent activation of Smad3 [17]. Epithelial-mesenchymal transition (EMT) of tubular epithelial cells has long been considered a pro-fibrotic mechanism. Recent evidence indicates that renal epithelial cells undergo a partial-EMT where epithelial cells develop fibroblast-like characteristics, without a complete conversion to fibroblasts [18]. IS induced an EMT-like process in vitro and in vivo through activation of the renin-angiotensin system (RAS), further contributing to renal fibrosis [19]. Moreover, IS induces renal expression of intercellular adhesion molecule 1 (ICAM-1), which is associated with monocyte infiltration into the kidney [20], and monocyte chemoattractant protein (MCP-1), a chemotactic cytokine implicated in macrophage recruitment and activation in tubulointerstitial inflammation [21].

Figure 2. Overview of putative mechanisms of renal toxicity for IS and pCS. Through various inflammatory and fibrotic pathways, both IS and pCS have been described to mediate toxicity to renal tubular cells in similar ways. Increased expression of various fibrotic genes such as TGF-β1, TIMP-1, pro-α1 (I) collagen contribute to alterations in the tubular cell morphology and structure of the ECM. Involvement of ROS and NF-κB reduce the proliferative capacity of tubular cells, contribute to partial EMT leading to fibrosis, and recruit macrophages to produce additional tubulointerstitial inflammation. Image created with BioRender.com.
The expression of Klotho, an anti-aging gene with renoprotective properties [22], is reduced in CKD patients [23]. IS was shown to induce Klotho depletion in vitro and in vivo [24], which has been linked with increased activation of NF-κB [25]. IS-induced Klotho depletion may be facilitated epigenetically through the CpG hypermethylation of the Klotho gene [26].
2.1.2. Mechanisms for the Progression of CVD
IS is also implicated with CVD and has clinically been correlated with mortality and comorbidities such as vascular calcification, vascular stiffness, and congestive heart failure in patients with ESRD [27]. IS is proposed to have detrimental effects on both cardiac cells and the vasculature (Figure 3). In vitro studies have shown that IS may play a role in endothelial dysfunction – an early marker of atherosclerosis [27]. In human umbilical vein endothelial cells (HUVECs), IS increased ROS production, decreased nitric oxide (NO) production and cell viability, and induced mRNA expression of NADPH oxidase 4 (Nox4) [28]. IS upregulates the expression of MCP-1 [29] and cell adhesion molecules E-selectin [30] and ICAM-1 [29], enhancing leukocyte interaction with endothelial cells and facilitating inflammation.
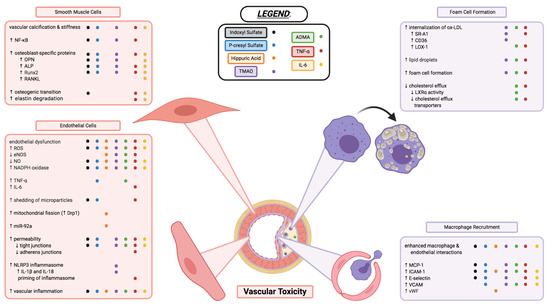
Figure 3. Vascular toxicity of uremic toxins through impacts on endothelial cell function, vascular smooth muscle cell morphology, and macrophage recruitment and transformation into foam cells of atherosclerotic plaques. Each coloured dot (see below and legend in figure) represents a uremic toxin having evidence in the literature to contribute to the various processes, or changes in cell activity and function described. Indoxyl sulfate (black), p-cresyl sulfate (blue), hippuric acid (orange), TMAO (purple), ADMA (green), TNF-α (red), and IL-6 (yellow) are shown to have varying effects in the cell types and cellular processes described above. Endothelial dysfunction and inflammation, vascular calcification and stiffness, enhanced interactions between macrophages and endothelial cells, as well as formation of foam cells are critical components to the various uremic toxins’ vascular toxicities. Some toxins may contribute to these mechanisms of toxicity but have yet to be elucidated or confirmed in the literature; some toxins have contradicting evidence that needs further investigation. This figure does not necessarily describe the complex interplay between the uremic toxins, inflammatory markers, and immune system that likely play a large interconnected role in the vascular toxicity contributing to CVD. Additional information on uremic toxins shown in this figure but not covered in this entry can be found in the full article: 10.3390/toxins13020142. Image created with BioRender.com.
Another common vascular complication in CKD is vascular calcification (Figure 3), which increases in prevalence as kidney function declines [31]. Vascular calcification is a risk factor for cardiovascular mortality and morbidity [31]. The differentiation of vascular smooth muscle cells (VSMCs) from a contractile to osteogenic phenotype is believed to be a key process in the development of vascular calcification in CKD. In both in vitro [32] and in vivo [33] experiments, IS has been shown to increase the expression of osteoblast-specific proteins, including runt-related transcription factor 2 (Runx2) and osteopontin (OPN). The activation of a PI3K/Akt/NF-κB pathway was shown to play a potential role in the osteogenic effects of IS [34].
IS also has direct pro-hypertrophic, pro-fibrotic, and pro-inflammatory effects on cardiomyocytes and cardiac fibroblasts. IS stimulated hypertrophy of neonatal rat cardiomyocytes, collagen synthesis in neonatal rat cardiac fibroblasts, and increased mRNA expression of pro-inflammatory cytokines in THP-1 cells (immortalized monocyte-like cell line), ultimately contributing to cardiac remodeling. These effects were proposed to be mediated through the activation of mitogen-activated protein kinase (MAPK) and NF-κB pathways [35]. Furthermore, cardiomyocytes treated with IS exhibited increased oxidative stress and reduced expression of UCP2, a member of the mitochondrial uncoupling proteins family with cardio-protective properties against ROS [36]. Restoring UCP2 in cardiomyocytes conferred protection against IS-induced oxidative stress and UCP2 downregulation [36].
2.2. p-Cresyl Sulfate
Metabolism of tyrosine and phenylalanine by intestinal bacteria yields a number of phenol derivatives, one of which is p-cresol [37]. P-cresol is absorbed and subsequently sulfated in the liver by SULT1A1 to produce p-cresyl sulfate (pCS) [38] (Figure 1). In vivo, more than 95% of p-cresol circulates as pCS [39]. Like IS, pCS is highly bound (90%) to albumin and is poorly cleared by dialysis [40] (Table 1). pCS is mainly cleared from the body by tubular secretion facilitated by basolateral uptake by OATs, namely OAT1/3 [41].
2.2.1. Mechanisms for the Progression of CKD
The accumulation of pCS shows similar toxic effects to IS. Much like IS, pCS induces oxidative stress and renal fibrosis/inflammation, and clinical studies have associated pCS with CKD progression [37] (Figure 2). pCS-treated HK-2 cells showed decreased viability, increased NADPH oxidase activity, and increased mRNA expression of TGF-β1, TIMP-1, and pro-α1 (I) collagen [42]. Similarly, kidneys from nephrectomised rats had increased tubular degeneration and fibrosis, increased TGF-β1 production, increased superoxide production, and upregulation of NADPH oxidase activity and expression [42]. pCS administration to nephrectomised rats resulted in CpG hypermethylation of the Klotho gene and reduced expression of Klotho in renal tubular cells [26]. Like IS, pCS also induces an EMT-like process through the activation of the RAS pathway, and the fibrotic effects of pCS were attenuated by inhibition of the RAS pathway [19]. P-cresyl sulfate has been shown to inhibit efflux transporters MRP4 and BCRP in proximal tubule cells, which may cause intracellular accumulation of toxins, including p-cresyl sulfate itself [43].
2.2.2. Mechanisms for the Progression of CVD
In hemodialysis patients, serum pCS levels were reported to be higher in patients with carotid atherosclerotic plaque and positively correlated with increased total plaque area. Serum pCS levels were also independently associated with the incidence and progression of carotid atherosclerotic plaque [44] and a significant independent predictor of plaque burden in patients attending vascular prevention clinics [45].
pCS has been implicated in vascular inflammation, vascular calcification, and atherogenesis. Cultured human endothelial and aortic smooth muscle cells treated with pCS showed enhanced ROS production, increased NADPH oxidase expression [46], and increased expression of pro-inflammatory factors MCP-1 and TNF-α [44]. pCS also increased mRNA levels of osteoblast-specific proteins in HASMCs, including alkaline phosphatase (ALP) and OPN, indicating a potential role of pCS in vascular calcification [46]. A recent study demonstrated pCS-induced ROS induces the phosphorylation of JNK, p38, and ERK, subsequently leading to increased NF-κB mediated expression of Runx2 and ALP [47]. pCS was also shown to increase the expression of adhesion molecules E-selectin, ICAM-1, and vascular cell adhesion molecule 1 (VCAM-1), promoting leukocyte-endothelium interaction in both endothelial cells and nephrectomised apoE-/- mice [44].
2.3. Targets for Therapeutic Intervention—IS and pCS
The most extensively studied method for lowering serum levels of gut-derived uremic toxins is oral administration of the spherical carbon adsorbent AST-120. AST-120 adsorbs precursors of gut-derived uremic toxins, including indole and p-cresol, in the colon and prevents their absorption into the circulation. A number of studies have demonstrated the ability of AST-120 administration to reduce plasma concentration of IS and pCS in both animal models [48] and hemodialysis patients [49]. In vivo, AST-120 has shown reno- and cardio-protective effects against uremic toxins [50][51]. Clinically, the benefits of AST-120 remain controversial. Although AST-120 has been shown to slow the decline of GFR in some studies [52], the large international randomized controlled trials EPPIC-1/EPPIC-2 showed a lack of benefit in delaying the progression of CKD, based on the primary endpoints of doubling of serum creatinine and initiation of dialysis or transplantation [53]. However, eGFR, a secondary endpoint in the EPPIC trials, declined significantly less with AST-120 treatment than placebo in EPICC-2 and in a pooled analysis of both trials. Furthermore, upon post hoc analysis of the EPPIC trials, it was suggested that AST-120 may delay CKD progression in certain patient subgroups [54][55]. While this post hoc analysis is intriguing, the best available evidence suggests AST-120 does not slow progression of CKD and further studies may be warranted in specific patient subgroups.
In addition to decreased clearance of gut-derived uremic toxins, CKD patients have a greater abundance of intestinal bacteria that produce indole and p-cresol [56]. Manipulation of the gut microbiome through the administration of pre-, pro- and synbiotics has been investigated as a therapeutic strategy for reducing the production of gut-derived uremic toxins. Overall, in healthy subjects, CKD patients, and hemodialysis patients, pre-, pro-, and synbiotics are reported to have a positive benefit in reducing the production of IS and pCS [57][58]. Currently, there are multiple registered clinical trials with plans to further investigate the effects of probiotics (NCT04390347) and synbiotics (NCT04527640) on IS and pCS levels in the setting of kidney disease. Using small molecules for targeting the gut microbiome may be another method of manipulating synthesis of gut-derived uremic toxins. Isoquercitrin, a naturally occurring small molecule, was recently demonstrated to reduce indole production without having microbicidal activity or inhibiting tryptophanase activity. It was proposed that isoquercitrin suppressed indole production by reducing tryptophan transport into bacteria through inhibition of the bacterial electron transport chain protein complex I and weakening the proton motive force [59].
By a similar rationale, modification of diet is another strategy to reduce uremic toxin production by gut bacteria. As IS and pCS are the result of bacterial breakdown of tryptophan and tyrosine/phenylalanine, respectively, reducing the dietary intake of these amino acid precursors via protein restriction can reduce the production of these gut-derived uremic solutes. In a protein restriction study, healthy subjects receiving a low protein diet had lower plasma levels and 24-h urinary excretion of IS than those receiving a high protein diet [60]. A decreasing trend in plasma levels and 24-h urinary excretion of pCS was also observed with the low protein diet, although the difference was not statistically significant (p = 0.07) [60]. It was also demonstrated that the dietary protein to fiber ratio was associated with both total serum IS and pCS levels in CKD patients [61]. Although evidence exists for the beneficial effects of protein restriction, protein malnutrition is common in CKD patients and may be exacerbated by dietary protein restriction [62]. Many clinical trials are planning to investigate the efficacy of dietary modulation in kidney disease (i.e., NCT03959228, NCT04505462).
2.4. Hippuric Acid
Hippuric acid is a gut-derived, protein-bound uremic toxin elevated in CKD [2]. The gut microbiome converts dietary polyphenols into benzoic acid through multiple phenolic reaction pathways, which is subsequently converted into hippuric acid via conjugation with glycine in the liver or kidneys [63] by glycine-N-acyltransferase (GLYAT) [64]. In circulation, hippuric acid is approximately 34% bound to albumin [65] and clearance of hippuric acid by dialysis is 64% [66]. OATs may play a role in the excretion of hippuric acid by the kidney [67].
2.4.1. Mechanisms for the Progression of CKD
Although there is some evidence suggesting hippuric acid contributes to the progression of renal fibrosis, the mechanisms of toxicity are not as well defined as IS and pCS. Hippuric acid has been implicated in promoting renal fibrosis and endothelial dysfunction by inducing oxidative stress. A recently published study demonstrated that hippuric acid contributes to the progression of renal fibrosis by disrupting redox homeostasis [68]. In this study, incubation of HK-2 cells with hippuric acid resulted in expression of fibrosis markers and induced extracellular matrix (ECM) imbalance, increased ROS and Nox4 expression, and activated TGF-β/Smad signaling. Nuclear factor erythroid 2-related factor 2 (NRF2) is a transcription factor that regulates the expression of thiol molecules, antioxidants, and detoxifying enzymes [68]. The formation of a NRF2-KEAP1-CUL3 complex negatively regulates NRF2 by ubiquitinating it for degradation. Under oxidative stress, the NRF2-KEAP1-CUL3 complex is disrupted and NRF2 can translocate into the nucleus to drive expression of the antioxidant network. Hippuric acid was shown to decrease the expression of NRF2 and its downstream antioxidant enzymes, and increase oxidative stress. Treatment with an NRF2 activator alleviated the reductions in NRF2 and antioxidant activity, and NRF2 was therefore proposed to be a potential therapeutic target against hippuric acid induced fibrosis [68].
2.4.2. Mechanisms for the Progression of CVD
Hippuric acid has been reported to promote endothelial dysfunction in vitro and in vivo, via generation of mitochondrial ROS. A shift in the balance between mitochondrial fusion and mitochondrial fission towards mitochondrial fission results in mitochondrial fragmentation and increased ROS generation. Treatment of human aortic endothelial cells (HAECs) with hippuric acid induced mitochondrial ROS production, reduced eNOS expression and increased expression of endothelial dysfunction markers ICAM-1 and von Willebrand factor (vWF). The mitochondria of HAECs treated with hippuric acid also exhibited morphological changes indicative of mitochondrial fragmentation and increased expression of Dynamin-related protein 1 (Drp1), a major regulator of mitochondrial fission. These effects on the endothelium were confirmed in vivo using nephrectomised CKD rats and healthy rats treated with hippuric acid. Collectively, these results suggest that hippuric acid promotes endothelial dysfunction at least partly by inducing mitochondrial fission and mitochondrial ROS production [69].
Another mechanism of hippuric acid-induced endothelial dysfunction may be through induction of miR-92a in endothelial cells. Hippuric acid was shown to induce miR-92a, a microRNA induced by oxidative stress in endothelial cells. miR-92a has been implicated in the angiogenic and atherosclerotic process and is increased in patients with CKD and pre-clinical models of CKD [70].
In a clinical study involving 80 hemodialysis patients, hippuric acid was associated with left ventricular hypertrophy (LVH), and hemodialysis patients with LVH had higher median pre-dialysis serum hippuric acid levels [71].
This entry is adapted from the peer-reviewed paper 10.3390/toxins13020142
References
- Meyer, T.W.; Hostetter, T.H. Uremia. N. Engl. J. Med. 2007, 357, 1316–1325.
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270.
- Madero, M.; Cano, K.B.; Campos, I.; Tao, X.; Maheshwari, V.; Brown, J.; Cornejo, B.; Handelman, G.; Thijssen, S.; Kotanko, P. Removal of protein-bound uremic toxins during hemodialysis using a binding competitor. Clin. J. Am. Soc. Nephrol. 2019, 14, 394–402.
- Leong, S.C.; Sirich, T.L. Indoxyl sulfate-review of toxicity and therapeutic strategies. Toxins 2016, 8, 358.
- Banoglu, E.; Jha, G.G.; King, R.S. Hepatic microsomal metabolism of indole to indoxyl, a precursor of indoxyl sulfate. Eur. J. Drug Metab. Pharmacokinet. 2001, 26, 235–240.
- Banoglu, E.; King, R.S. Sulfation of indoxyl by human and rat aryl (phenol) sulfotransferases to form indoxyl sulfate. Eur. J. Drug Metab. Pharmacokinet. 2002, 27, 135–140.
- Enomoto, A.; Takeda, M.; Tojo, A.; Sekine, T.; Cha, S.H.; Khamdang, S.; Takayama, F.; Aoyama, I.; Nakamura, S.; Endou, H.; et al. Role of organic anion transporters in the tubular transport of indoxyl sulfate and the induction of its nephrotoxicity. J. Am. Soc. Nephrol. 2002, 13, 1711–1720.
- Devine, E.; Krieter, D.H.; Rüth, M.; Jankovski, J.; Lemke, H.D. Binding affinity and capacity for the uremic toxin indoxyl sulfate. Toxins 2014, 6, 416–430.
- Fagugli, R.M.; De Smet, R.; Buoncristiani, U.; Lameire, N.; Vanholder, R. Behavior of non-protein-bound and protein-bound uremic solutes during daily hemodialysis. Am. J. Kidney Dis. 2002, 40, 339–347.
- Fujii, H.; Goto, S.; Fukagawa, M. Role of uremic toxins for kidney, cardiovascular, and bone dysfunction. Toxins 2018, 10, 202.
- Stubbs, J.R.; House, J.A.; Ocque, A.J.; Zhang, S.; Johnson, C.; Kimber, C.; Schmidt, K.; Gupta, A.; Wetmore, J.B.; Nolin, T.D.; et al. Serum Trimethylamine-N-Oxide is Elevated in CKD and Correlates with Coronary Atherosclerosis Burden. J. Am. Soc. Nephrol. 2016, 27, 305–313.
- Sitar, M.E.; Kayacelebi, A.A.; Beckmann, B.; Kielstein, J.T.; Tsikas, D. Asymmetric dimethylarginine (ADMA) in human blood: Effects of extended haemodialysis in the critically ill patient with acute kidney injury, protein binding to human serum albumin and proteolysis by thermolysin. Amino Acids 2015, 47, 1983–1993.
- Jacobi, J.; Tsao, P.S. Asymmetrical Dimethylarginine in Renal Disease: Limits of Variation or Variation Limits? Am. J. Nephrol. 2008, 28, 224–237.
- Motojima, M.; Hosokawa, A.; Yamato, H.; Muraki, T.; Yoshioka, T. Uremic toxins of organic anions up-regulate PAI-1 expression by induction of NF-κB and free radical in proximal tubular cells. Kidney Int. 2003, 63, 1671–1680.
- Owada, S.; Goto, S.; Bannai, K.; Hayashi, H.; Nishijima, F.; Niwa, T. Indoxyl sulfate reduces superoxide scavenging activity in the kidneys of normal and uremic rats. Am. J. Nephrol. 2008, 28, 446–454.
- Shimizu, H.; Bolati, D.; Adijiang, A.; Muteliefu, G.; Enomoto, A.; Nishijima, F.; Dateki, M.; Niwa, T. NF-κb plays an important role in indoxyl sulfate-induced cellular senescence, fibrotic gene expression, and inhibition of proliferation in proximal tubular cells. Am. J. Physiol. Cell Physiol. 2011, 301, 1201–1212.
- Shimizu, H.; Yisireyili, M.; Nishijima, F.; Niwa, T. Indoxyl sulfate enhances p53-tgf-β1-smad3 pathway in proximal tubular cells. Am. J. Nephrol. 2013, 37, 97–103.
- Sheng, L.; Zhuang, S. New Insights Into the Role and Mechanism of Partial Epithelial-Mesenchymal Transition in Kidney Fibrosis. Front. Physiol. 2020, 11, 1–11.
- Sun, C.Y.; Chang, S.C.; Wu, M.S. Uremic toxins induce kidney fibrosis by activating intrarenal renin-angiotensin-aldosterone system associated epithelial-to-mesenchymal transition. PLoS ONE 2012, 7, 1–10.
- Miyazaki, T.; Ise, M.; Seo, H.; Niwa, T. Indoxyl sulfate increases the gene expressions of TGF-beta 1, TIMP-1 and pro-alpha 1(I) collagen in uremic rat kidneys. Kidney Int. Suppl. 1997, 62, S15–S22.
- Shimizu, H.; Bolati, D.; Higashiyama, Y.; Nishijima, F.; Shimizu, K.; Niwa, T. Indoxyl sulfate upregulates renal expression of MCP-1 via production of ROS and activation of NF-κB, p53, ERK, and JNK in proximal tubular cells. Life Sci. 2012, 90, 525–530.
- Haruna, Y.; Kashihara, N.; Satoh, M.; Tomita, N.; Namikoshi, T.; Sasaki, T.; Fujimori, T.; Xie, P.; Kanwar, Y.S. Amelioration of progressive renal injury by genetic manipulation of Klotho gene. Proc. Natl. Acad. Sci. USA 2007, 104, 2331–2336.
- Koh, N.; Fujimori, T.; Nishiguchi, S.; Tamori, A.; Shiomi, S.; Nakatani, T.; Sugimura, K.; Kishimoto, T.; Kinoshita, S.; Kuroki, T.; et al. Severely reduced production of klotho in human chronic renal failure kidney. Biochem. Biophys. Res. Commun. 2001, 280, 1015–1020.
- Shimizu, H.; Bolati, D.; Adijiang, A.; Adelibieke, Y.; Muteliefu, G.; Enomoto, A.; Higashiyama, Y.; Higuchi, Y.; Nishijima, F.; Niwa, T. Indoxyl sulfate downregulates renal expression of Klotho through production of ros and activation of nuclear factor-κB. Am. J. Nephrol. 2011, 33, 319–324.
- Zhao, Y.; Banerjee, S.; Dey, N.; LeJeune, W.S.; Sarkar, P.S.; Brobey, R.; Rosenblatt, K.P.; Tilton, R.G.; Choudhary, S. Klotho depletion contributes to increased inflammation in kidney of the db/db mouse model of diabetes via RelA (serine)536 phosphorylation. Diabetes 2011, 60, 1907–1916.
- Sun, C.Y.; Chang, S.C.; Wu, M.S. Suppression of Klotho expression by protein-bound uremic toxins is associated with increased DNA methyltransferase expression and DNA hypermethylation. Kidney Int. 2012, 81, 640–650.
- Hung, S.; Kuo, K.; Wu, C.; Tarng, D. Indoxyl Sulfate: A Novel Cardiovascular Risk Factor in Chronic Kidney Disease. J. Am. Heart Assoc. 2017, 6, e005022.
- Tumur, Z.; Niwa, T. Indoxyl sulfate inhibits nitric oxide production and cell viability by inducing oxidative stress in vascular endothelial cells. Am. J. Nephrol. 2009, 29, 551–557.
- Tumur, Z.; Shimizu, H.; Enomoto, A.; Miyazaki, H.; Niwa, T. Indoxyl sulfate upregulates expression of ICAM-1 and MCP-1 by oxidative stress-induced NF-κB activation. Am. J. Nephrol. 2010, 31, 435–441.
- Ito, S.; Osaka, M.; Higuchi, Y.; Nishijima, F.; Ishii, H.; Yoshida, M. Indoxyl sulfate induces leukocyte-endothelial interactions through up-regulation of E-selectin. J. Biol. Chem. 2010, 285, 38869–38875.
- Zou, D.; Wu, W.; He, Y.; Ma, S.; Gao, J. The role of klotho in chronic kidney disease. BMC Nephrol. 2018, 19, 1–12.
- Muteliefu, G.; Enomoto, A.; Jiang, P.; Takahashi, M.; Niwa, T. Indoxyl sulphate induces oxidative stress and the expression of osteoblast-specific proteins in vascular smooth muscle cells. Nephrol. Dial. Transplant. 2009, 24, 2051–2058.
- Adijiang, A.; Goto, S.; Uramoto, S.; Nishijima, F.; Niwa, T. Indoxyl sulphate promotes aortic calcification with expression of osteoblast-specific proteins in hypertensive rats. Nephrol. Dial. Transplant. 2008, 23, 1892–1901.
- He, X.; Jiang, H.; Gao, F.; Liang, S.; Wei, M.; Chen, L. Indoxyl sulfate-induced calcification of vascular smooth muscle cells via the PI3K/Akt/NF-κB signaling pathway. Microsc. Res. Tech. 2019, 82, 2000–2006.
- Lekawanvijit, S.; Adrahtas, A.; Kelly, D.J.; Kompa, A.R.; Wang, B.H.; Krum, H. Does indoxyl sulfate, a uraemic toxin, have direct effects on cardiac fibroblasts and myocytes? Eur. Heart J. 2010, 31, 1771–1779.
- Yang, K.; Xu, X.; Nie, L.; Xiao, T.; Guan, X.; He, T.; Yu, Y.; Liu, L.; Huang, Y.; Zhang, J.; et al. Indoxyl sulfate induces oxidative stress and hypertrophy in cardiomyocytes by inhibiting the AMPK/UCP2 signaling pathway. Toxicol. Lett. 2015, 234, 110–119.
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Glorieux, G. p-Cresyl Sulfate. Toxins 2017, 9, 52.
- Gamage, N.; Barnett, A.; Hempel, N.; Duggleby, R.G.; Windmill, K.F.; Martin, J.L.; McManus, M.E. Human Sulfotransferases and Their Role in Chemical Metabolism. Toxicol. Sci. 2006, 90, 5–22.
- Meijers, B.K.I.; Van kerckhoven, S.; Verbeke, K.; Dehaen, W.; Vanrenterghem, Y.; Hoylaerts, M.F.; Evenepoel, P. The Uremic Retention Solute p-Cresyl Sulfate and Markers of Endothelial Damage. Am. J. Kidney Dis. 2009, 54, 891–901.
- Martinez, A.W.; Recht, N.S.; Hostetter, T.H.; Meyer, T.W. Removal of P-Cresol Sulfate by Hemodialysis. J. Am. Soc. Nephrol. 2005, 16, 3430–3436.
- Miyamoto, Y.; Watanabe, H.; Noguchi, T.; Kotani, S.; Nakajima, M.; Kadowaki, D.; Otagiri, M.; Maruyama, T. Organic anion transporters play an important role in the uptake of p-cresyl sulfate, a uremic toxin, in the kidney. Nephrol. Dial. Transplant. 2011, 26, 2498–2502.
- Watanabe, H.; Miyamoto, Y.; Honda, D.; Tanaka, H.; Wu, Q.; Endo, M.; Noguchi, T.; Kadowaki, D.; Ishima, Y.; Kotani, S.; et al. P-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int. 2013, 83, 582–592.
- Mutsaers, H.A.M.; Caetano-Pinto, P.; Seegers, A.E.M.; Dankers, A.C.A.; van den Broek, P.H.H.; Wetzels, J.F.M.; van den Brand, J.A.J.G.; van den Heuvel, L.P.; Hoenderop, J.G.; Wilmer, M.J.G.; et al. Proximal tubular efflux transporters involved in renal excretion of p-cresyl sulfate and p-cresyl glucuronide: Implications for chronic kidney disease pathophysiology. Toxicol. Vitr. 2015, 29, 1868–1877.
- Jing, Y.J.; Ni, J.W.; Ding, F.H.; Fang, Y.H.; Wang, X.Q.; Wang, H.B.; Chen, X.N.; Chen, N.; Zhan, W.W.; Lu, L.; et al. P-Cresyl sulfate is associated with carotid arteriosclerosis in hemodialysis patients and promotes atherogenesis in apoE-/- mice. Kidney Int. 2016, 89, 439–449.
- Bogiatzi, C.; Gloor, G.; Allen-Vercoe, E.; Reid, G.; Wong, R.G.; Urquhart, B.L.; Dinculescu, V.; Ruetz, K.N.; Velenosi, T.J.; Pignanelli, M.; et al. Metabolic products of the intestinal microbiome and extremes of atherosclerosis. Atherosclerosis 2018, 273, 91–97.
- Watanabe, H.; Miyamoto, Y.; Enoki, Y.; Ishima, Y.; Kadowaki, D.; Kotani, S.; Nakajima, M.; Tanaka, M.; Matsushita, K.; Mori, Y.; et al. p-Cresyl sulfate, a uremic toxin, causes vascular endothelial and smooth muscle cell damages by inducing oxidative stress. Pharmacol. Res. Perspect. 2015, 3, 1–12.
- Chang, J.F.; Hsieh, C.Y.; Liou, J.C.; Liu, S.H.; Hung, C.F.; Lu, K.C.; Lin, C.C.; Wu, C.C.; Ka, S.M.; Wen, L.L.; et al. Scavenging intracellular ros attenuates p-cresyl sulfate-triggered osteogenesis through mapk signaling pathway and NF-κB activation in human arterial smooth muscle cells. Toxins 2020, 12, 472.
- Velenosi, T.J.; Hennop, A.; Feere, D.A.; Tieu, A.; Kucey, A.S.; Kyriacou, P.; McCuaig, L.E.; Nevison, S.E.; Kerr, M.A.; Urquhart, B.L. Untargeted plasma and tissue metabolomics in rats with chronic kidney disease given AST-120. Sci. Rep. 2016, 6, 1–12.
- Yamamoto, S.; Kazama, J.J.; Omori, K.; Matsuo, K.; Takahashi, Y.; Kawamura, K.; Matsuto, T.; Watanabe, H.; Maruyama, T.; Narita, I. Continuous Reduction of Protein-Bound Uraemic Toxins with Improved Oxidative Stress by Using the Oral Charcoal Adsorbent AST-120 in Haemodialysis Patients. Sci. Rep. 2015, 5, 3–10.
- Nakagawa, N.; Hasebe, N.; Sumitomo, K.; Fujino, T.; Fukuzawa, J.; Hirayama, T.; Kikuchi, K. An oral adsorbent, AST-120, suppresses oxidative stress in uremic rats. Am. J. Nephrol. 2006, 26, 455–461.
- Yamamoto, S.; Zuo, Y.; Ma, J.; Yancey, P.G.; Hunley, T.E.; Motojima, M.; Fogo, A.B.; Linton, M.F.; Fazio, S.; Ichikawa, I.; et al. Oral activated charcoal adsorbent (AST-120) ameliorates extent and instability of atherosclerosis accelerated by kidney disease in apolipoprotein E-deficient mice. Nephrol. Dial. Transplant. 2011, 26, 2491–2497.
- Asai, M.; Kumakura, S.; Kikuchi, M. Review of the efficacy of AST-120 (KREMEZIN®) on renal function in chronic kidney disease patients. Ren. Fail. 2019, 41, 47–56.
- Schulman, G.; Berl, T.; Beck, G.J.; Remuzzi, G.; Ritz, E.; Arita, K.; Kato, A.; Shimizu, M. Randomized Placebo-Controlled EPPIC Trials of AST-120 in CKD. J. Am. Soc. Nephrol. 2015, 26, 1732–1746.
- Schulman, G.; Berl, T.; Beck, G.J.; Remuzzi, G.; Ritz, E.; Shimizu, M.; Kikuchi, M.; Shobu, Y. Risk factors for progression of chronic kidney disease in the EPPIC trials and the effect of AST-120. Clin. Exp. Nephrol. 2018, 22, 299–308.
- Schulman, G.; Berl, T.; Beck, G.J.; Remuzzi, G.; Ritz, E.; Shimizu, M.; Shobu, Y.; Kikuchi, M. The effects of AST-120 on chronic kidney disease progression in the United States of America: A post hoc subgroup analysis of randomized controlled trials. BMC Nephrol. 2016, 17, 141.
- Wong, J.; Piceno, Y.M.; DeSantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of Urease- and Uricase-Containing, Indole- and p-Cresol-Forming and Contraction of Short-Chain Fatty Acid-Producing Intestinal Microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237.
- Rossi, M.; Klein, K.; Johnson, D.W.; Campbell, K.L. Pre-, pro-, and synbiotics: Do they have a role in reducing uremic toxins? a systematic review and meta-analysis. Int. J. Nephrol. 2012, 2012.
- Rossi, M.; Johnson, D.W.; Morrison, M.; Pascoe, E.M.; Coombes, J.S.; Forbes, J.M.; Szeto, C.C.; McWhinney, B.C.; Ungerer, J.P.J.; Campbell, K.L. Synbiotics easing renal failure by improving gut microbiology (SYNERGY): A randomized trial. Clin. J. Am. Soc. Nephrol. 2016, 11, 223–231.
- Wang, Y.; Li, J.; Chen, C.; Lu, J.; Yu, J.; Xu, X.; Peng, Y.; Zhang, S.; Jiang, S.; Guo, J.; et al. Targeting the gut microbial metabolic pathway with small molecules decreases uremic toxin production. Gut Microbes 2020, 12, 1–19.
- Poesen, R.; Mutsaers, H.A.M.; Windey, K.; Van Den Broek, P.H.; Verweij, V.; Augustijns, P.; Kuypers, D.; Jansen, J.; Evenepoel, P.; Verbeke, K.; et al. The influence of dietary protein intake on mammalian tryptophan and phenolic metabolites. PLoS ONE 2015, 10, 1–12.
- Rossi, M.; Johnson, D.W.; Xu, H.; Carrero, J.J.; Pascoe, E.; French, C.; Campbell, K.L. Dietary protein-fiber ratio associates with circulating levels of indoxyl sulfate and p-cresyl sulfate in chronic kidney disease patients. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 860–865.
- Bammens, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. Evidence for impaired assimilation of protein in chronic renal failure. Kidney Int. 2003, 64, 2196–2203.
- Pallister, T.; Jackson, M.A.; Martin, T.C.; Zierer, J.; Jennings, A.; Mohney, R.P.; MacGregor, A.; Steves, C.J.; Cassidy, A.; Spector, T.D.; et al. Hippurate as a metabolomic marker of gut microbiome diversity: Modulation by diet and relationship to metabolic syndrome. Sci. Rep. 2017, 7, 1–9.
- Badenhorst, C.P.S.; van der Sluis, R.; Erasmus, E.; van Dijk, A.A. Glycine conjugation: Importance in metabolism, the role of glycine N -acyltransferase, and factors that influence interindividual variation. Expert Opin. Drug Metab. Toxicol. 2013, 9, 1139–1153.
- Deltombe, O.; Van Biesen, W.; Glorieux, G.; Massy, Z.; Dhondt, A.; Eloot, S. Exploring protein binding of uremic toxins in patients with different stages of chronic kidney disease and during hemodialysis. Toxins 2015, 7, 3933–3946.
- Zaidi, N.; Ajmal, M.R.; Rabbani, G.; Ahmad, E.; Khan, R.H. A Comprehensive Insight into Binding of Hippuric Acid to Human Serum Albumin: A Study to Uncover Its Impaired Elimination through Hemodialysis. PLoS ONE 2013, 8.
- Deguchi, T.; Ohtsuki, S.; Otagiri, M.; Takanaga, H.; Asaba, H.; Mori, S.; Terasaki, T. Major role of organic anion transporter 3 in the transport of indoxyl sulfate in the kidney. Kidney Int. 2002, 61, 1760–1768.
- Sun, B.; Wang, X.; Liu, X.; Wang, L.; Ren, F.; Wang, X.; Leng, X. Hippuric acid promotes renal fibrosis by disrupting redox homeostasis via facilitation of NRF2–KEAP1–CUL3 interactions in chronic kidney disease. Antioxidants 2020, 9, 783.
- Huang, M.; Wei, R.; Wang, Y.; Su, T.; Li, P.; Chen, X. The uremic toxin hippurate promotes endothelial dysfunction via the activation of Drp1-mediated mitochondrial fission. Redox Biol. 2018, 16, 303–313.
- Shang, F.; Wang, S.-C.; Hsu, C.-Y.; Miao, Y.; Martin, M.; Yin, Y.; Wu, C.-C.; Wang, Y.-T.; Wu, G.; Chien, S.; et al. MicroRNA-92a Mediates Endothelial Dysfunction in CKD. J. Am. Soc. Nephrol. 2017, 28, 3251–3261.
- Yu, T.-H.; Tang, W.-H.; Lu, Y.-C.; Wang, C.-P.; Hung, W.-C.; Wu, C.-C.; Tsai, I.-T.; Chung, F.-M.; Houng, J.-Y.; Lan, W.-C.; et al. Association between hippuric acid and left ventricular hypertrophy in maintenance hemodialysis patients. Clin. Chim. Acta. 2018, 484, 47–51.