The tripartite motif (TRIM) family comprises at least 80 members in humans, with most having ubiquitin or SUMO E3 ligase activity conferred by their N-terminal RING domain. TRIMs regulate a wide range of processes in ubiquitination- or sumoylation-dependent manners in most cases, and fewer as adaptors. Their roles in the regulation of viral infections, autophagy, cell cycle progression, DNA damage and other stress responses, and carcinogenesis are being increasingly appreciated, and their E3 ligase activities are attractive targets for developing specific immunotherapeutic strategies for immune diseases and cancers.
- TRIMs
- ubiquitination
- PRR
- IFN-I
- IRFs
1. Introduction
In mammalians, interferons (IFNs) include three types, type I, II, and III. Type I IFNs (IFN-Is) include the majority of 26 isoforms of IFNα that are encoded by 13 genes, and one IFNβ that is encoded by the single gene IFNB, as well as other minor subtypes, including IFNε, IFNκ, IFNω, IFNδ, IFNτ, and IFNζ. IFNαs are mainly secreted by plasmacytoid dendritic cells (pDCs) and IFNβ is mainly secreted by fibroblasts. All IFN-Is signal through the integral membrane IFNAR1 and -2 heterodimer, and play crucial roles in the first line of innate immune response and subsequent adaptive immune response in response to viral or bacterial infections [1].
Importantly, recent studies have shown that IFN-Is play a dual role in chronic viral infections. At the early stage of infection, they have potent antiviral activity. However, at late stages, a low level of prolonged IFN-I signaling, exemplified by chronic infection of viruses such as HIV and HCV [2][3][4], triggers long-term chronic immune activation that proceeds to T cell exhaustion and inflammaging/immunosenescence in both direct and indirect manners [5][6] and therefore serves as a bridge that links innate and adaptive immune responses [4][5][7][8][9][10]. For example, the engagement of TLR7 in HIV-infected CD4+ T cells induces anergy/unresponsiveness, accounting for the impaired T cell function by chronic HIV infection [11]. A prolonged IFN-I response also facilitates the establishment of TME (tumor microenvironment) [4][5][7][12][13][14]. IFN-Is also play crucial roles in cellular development and homeostasis [5][6][15][16][17]. Aberrant production of IFN-Is is associated with many types of diseases, including autoimmune disorders and cancers [6][18][19][20]. Therefore, it is of fundamental importance to understand the precise mechanisms of how IFN-Is are regulated in different biological contexts [21][22].
Ubiquitination is a pervasive theme equally important to phosphorylation of proteins in myriad processes. Ubiquitin (Ub) is a 76-amino acid protein that is ubiquitously distributed and highly conserved throughout eukaryotic organisms. The Ub protein can be free or conjugated to a lysine site of a protein substrate through its 3’-end. This conjugation process involves E1 activating enzyme, E2 conjugating enzyme, and E3 ligase, with the E3 ligase determining the specificity of the substrate. Ub itself has seven internal lysine residues (K6, K11, K27, K29, K33, K48, and K63), and each can serve as the Ub target to link another Ub. If only a single Ub is conjugated to each lysine site of the substrates, it is called mono (also only one lysine site on the substrate) or multi (more than one lysine site on the substrate) ubiquitination. If the substrate is Ub itself, polyubiquitin chains will be formed on the substrate. Usually, a polyubiquitin chain contains more than 4 Ub molecules. In the last decade, non-canonical ubiquitination types on serine, threonine, and cysteine sites other than lysine site have been identified, and their importance in specific cellular functions has been recognized [23][24].
The most well-understood type of ubiquitination is K48-linked polyubiquitination, which is principally known as the major process whereby proteins are targeted for proteasomal degradation through the 26S proteasome. Later, nonproteolytic types of polyubiquitination (represented by K63-linked polyubiquitination), monoubiquitination, and linear ubiquitination have been gradually identified [25][26][27]. More recently, other ubiquitination-like modifications (e.g., sumoylation, acetylation, ISGylation, neddylation, palmitoylation, and UFMylation) have also been discovered. The roles of these posttranslational modifications (PTMs) in a myriad of cellular processes, such as receptor internalization (endocytosis), vesicle trafficking, immune response and inflammation, DNA damage response, autophagy, and cell death, have been greatly appreciated [27][28][29][30][31][32][33][34][35].
IFN-I production is controlled at multiple layers to ensure appropriate mounting of antiviral and antitumor immune responses. It is clear that both host and viral ubiquitin systems play pivotal roles in IFN-I-mediated innate immunity and in cellular transformation mediated by oncogenic viruses represented by EBV (Epstein-Barr Virus), KSHV (Kaposi’s sarcoma-associated herpesvirus), and HPV (human papillomavirus) [36][37][38][39][40][41].
2. PRR Signaling Pathways to IFN-I Production
IFN-Is are produced downstream of the signaling pathways of host germline-encoded pathogen recognition receptors (PRRs), which are expressed on the cell membrane or in the cytoplasm of the cells of the innate immune system, in response to pathogen-associated molecular patterns (PAMPs) that include pathogenic nucleic acids, LPS, and proteins, or in response to host damage-associated molecular patterns (DAMPs), such as self-nuclei acids, heat-shock proteins, and HMGB1. Recognition of PAMPs or DAMPs by PRRs triggers signal cascades that activate the transcription factors, including NFκB, Interferon regulatory factors (IRFs), and AP1, or activate caspase-mediated cell death and inflammation.
PRRs include the well-known transmembrane Toll-like receptors (TLRs) (Figure 1) and an increasing pool of “Toll-free” receptors [22]. Endosomal TLRs (TLR3, -7, -9, and murine TLR8) and endocytic TLR4, as well as cytoplasmic RIG-I and cGAS, amongst others, are able to recognize pathogenic or host cell nucleic acids (LPS for TLR4) to activate IRFs in addition to NFκB and AP1, which induce IFN-Is and also pro-inflammatory cytokines [42]. Self-nucleic acids are derived from the nucleus or mitochondria of the cells suffering from endogenous or exogenous stresses, such as DNA replication, oxidative stress, DNA damage, and cell death [43][44][45][46][47]. While the transcription of IFNαs is solely dependent on IRFs, full transactivation of the IFNβ promoter requires the cooperation of IRFs, NFκB, and other co-factors in the transcriptional complex named enhanceosome [48].
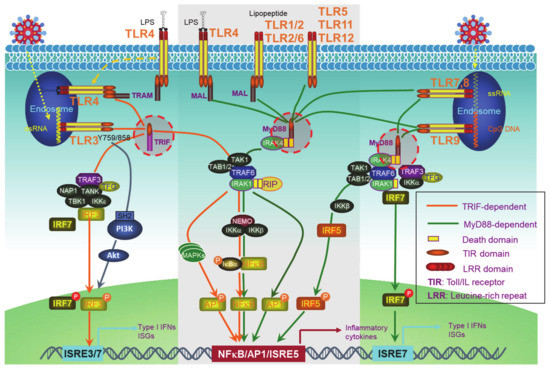
Figure 1. Toll-like receptor (TLR)signaling pathways. The TLR family has 13 members, among which, endosomal TLR3, -7, -9, and murine TLR8, and endocytic TLR4 are able to trigger signaling for IFN-I production. TLRs that cannot trigger the activation of Interferon regulatory factors (IRFs) (The middle part in the gray frame) do not contribute to IFN-I production. All the TLRs have a TIR domain in the cytoplasm, which recruits adaptor proteins also with a TIR domain at their C-terminus. TRIF and MyD88, two adaptor proteins, bridge all TLRs to downstream signaling molecules, leading to the activation of NFκB, IRFs, and AP1. IRF1, -3, -5, -7, and -8 are the transcription factors for both IFNα and IFNβ transcription in different cell contexts, but a full IFNβ transcription requires the enhanceosome complex that contains NFκB, IRF3, -7, ATF-2/c-Jun, and HMGIY (high mobility group I(Y)). ISRE: Interferon-stimulated response element. ISRE3/7: ISRE that binds to IRF3//7. ISRE7: ISRE that binds to IRF7.
3. The TRIM Family
The tripartite motif (TRIM) family of proteins is large and includes at least 80 members in humans, with most having E3 ligase activity for target-specific ubiquitination, and plays crucial roles in innate immunity, transcription, autophagy, and carcinogenesis [49][50]. The N-terminal TRIM motif includes the conserved RBCC domain that comprises of three subdomains: 1 RING domain that confers with E3 ligase activity (8 human TRIMs do not have the RING domain), 0~2 B-box ZNF domains (B1+B2 or B2 alone), and 0~1 coil–coil region that is associated with B-boxes. According to the diversity of the C-terminuses and genomic organization, TRIM proteins are grouped into Group1 and Group 2. Members in Group 1 possess a variety of C-terminal domains (COS, FN3, ACID, PRY, PHD-BROMO, FIL, NHL, MATH, ARF, and TM) and exist in both vertebrate and invertebrates, and those in Group 2 possess a C-terminal SPRY domain, and they are absent in invertebrates (Figure 2) [49][50][51]. The SPY-SPRY domain is critical for TRIM proteins’ interaction with their substrates.
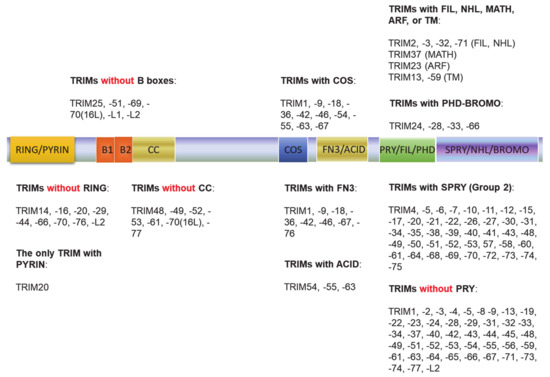
Figure 2. The tripartite motif (TRIM) family protein domain alignment. The TRIM family includes at least 80 members in humans. The N-terminal TRIM motif includes the conserved RBCC domain that comprises of three subdomains: 1 RING domain that confers with E3 ligase activity (8 TRIMs in humans do not have the RING domain), 0~2 B-box ZNF domains (B1+B2 or B2 alone), and 0~1 coil–coil region that is associated with B-boxes. According to the diversity of the C-terminuses and genomic organization, TRIM proteins are grouped into Group1 and Group 2. Members in Group 1 possess a variety of C-terminal domains (COS, FN3, ACID, PRY, PHD-BROMO, FIL, NHL, MATH, ARF, and TM) and exist in both vertebrate and invertebrates, and those in Group 2 possess a C-terminal SPRY domain, and they are absent in invertebrates.
Many TRIMs are inducible by IFN-Is, and play crucial roles in IFN-I-mediated innate immune regulation, with the involvement of ubiquitination and sumoylation in most cases [50][52][53][54][55][56][57][58]. These TRIMs can target most if not all components of the PRR and Jak-STAT1 IFN-I pathways, including different ligands (PAMPs and DAMPs); the receptors such as TLRs, cGAS, and DDX41; the adaptors MyD88, TRIF, STING, and TRAF6 and -3; the kinases IKKs and TAK1; and the transcription factors IRF3 and -7 and NFκB (Table 1). TRIM genes evolve parallelly with the immune system, further supporting their roles as regulators of immune responses [51][53].
Table 1. TRIMs in the regulation of Interferon (IFN)-I-mediated innate immune network.
|
TRIM |
Synonym |
Targets in the IFN-I Network |
Ub Conjugation Type |
Outcome of Conjugation |
Selected References |
|---|---|---|---|---|---|
|
TRIM3 |
RNF97 |
TLR3 |
K63 |
Promotes ESCRT-mediated TLR3 sorting to endosomes |
[59] |
|
TRIM4 |
RNF87 |
RIG-I |
K63 |
Activation |
[60] |
|
TRIM5α |
RNF88 |
HIV Gag |
K48 |
Degradation |
|
|
TAK1 |
K63 |
Activation |
[63] |
||
|
TRIM12c in mice |
TRAF6 |
K63 (?) |
Activation |
[62] |
|
|
TRIM6 |
RNF89 |
Ebola VP35 |
Poly |
Promotes VP35 IFN-I inhibitory activity |
[64] |
|
IKKε |
Free K48 |
Activation of IKKε, leading to STAT1 activation |
[65] |
||
|
TRIM7 |
RNF90 GNIP |
Zika virus envelope (E) |
K63 |
Enhances virus attachment and entry into the cell |
[66] |
|
TLR4 |
NA* |
Promotes TLR4 activation |
[67] |
||
|
TRIM8 |
RNF27 GERP |
TRIF |
K6, K33 |
Disrupts the TRIF-TBK1 complex |
[68] |
|
TAK1 |
K63 |
Activation |
|||
|
IRF7 |
Protects p-IRF7 from Pin1-mediated proteasomal degradation in the nucleus |
[71] |
|||
|
SOCS1 |
K48 (?) |
Degradation |
[72] |
||
|
PIAS3 |
K48 |
Degradation |
[73] |
||
|
Interaction (?) |
Promotes PIAS3 nucleus-to-cytoplasm translocation |
[74] |
|||
|
TRIM9s |
RNF91 SPRING |
TBK1 |
Interaction |
Recruits GSK3β and TBK1, leading to TBK1 activation |
[75] |
|
TRIM9 |
β-TrCP |
Interaction |
Stabilizes IκBα |
||
|
TRIM11 |
RNF92 BIA1 |
TBK1 |
Interaction |
Inhibits TBK1 activation |
[77] |
|
TRIM5 |
NA* |
Degradation |
[78] |
||
|
TRIM13 |
RNF77 RFP2 CAR LEU5 DLEU5 |
RIG-I |
Interaction |
Potentiates RIG-I activity |
[79] |
|
MDA5 |
Interaction |
Inhibition |
[79] |
||
|
TRAF6 |
K29 |
Activation |
[80] |
||
|
NEMO |
K48 |
Degradation |
[81] |
||
|
TRIM14 |
KIAA0129 |
HCV NS5A |
K48 (?) |
Degradation |
[82] |
|
cGAS, TBK1 |
Interaction |
Inhibition of autophagic degradation of cGAS |
|||
|
MAVS |
Interaction |
Recruitment of NEMO to MAVS signalosome |
[84] |
||
|
TRIM15 |
RNF93 ZNF178 ZNFB7 |
MAVS |
NA* |
Promotes RIG-I-mediated IFN production |
[86] |
|
TRIM19 |
RNF71 PML MYL |
HIV genome |
Sequestrates HIV genome in the cytoplasm, blocking HIV transduction |
[87] |
|
|
HFV Tas |
Represses HFV transcription by preventing Tas binding to viral DNA |
[88] |
|||
|
LCMV Z |
Inhibits LCMV replication |
[89] |
|||
|
hCMV IE1 |
Interaction |
IE1 forms a complex with TRIM19-STAT1/2 to impede IFN-I signaling |
[90] |
||
|
STAT1/2 |
Induction and stabilization, promoting IFN-I signaling |
[90] |
|||
|
Pin1 (by TRIM19IV) |
Regulates the cellular distribution of Pin1 |
[91] |
|||
|
Ubc9 (The only SUMO E2) |
Required for IFN-induced global sumoylation |
[92] |
|||
|
NFκB |
Inhibits NFκB-mediated transcription and survival |
[93] |
|||
|
Promotes IKKε-mediated p65 phosphorylation and NFκB activity |
[94] |
||||
|
ROS |
Functions as an ROS sensor promoting p53 activation |
[95] |
|||
|
TRIM20 |
Pyrin MEFV |
p65 |
Interaction |
Promotes p65 nuclear translocation |
[96] |
|
IκBα |
Promotes IκBα degradation |
[96] |
|||
|
TRIM21 |
RNF81 Ro52 SSA1 |
DDX41 |
K48 |
Degradation |
[97] |
|
MAVS |
K27 |
Activation |
[98] |
||
|
FADD |
Interaction |
Promotes IRF7 ubiquitination-mediated degradation |
[99] |
||
|
TAK1 |
Free K63 |
Activates TAK1, leading to the activation of NFκB, AP1, and IRFs |
|||
|
IKKβ |
Mono-Ub |
Autophagic degradation |
[102] |
||
|
IRF3 |
Interaction |
Protects p-IRF3 from Pin1-mediated proteasomal degradation |
[103] |
||
|
K48 |
Targets IRF3 for proteosomal degradation |
||||
|
Interacts with ULK1, Beclin1, and p62 |
Targets IRF3 for autophagic degradation |
[106] |
|||
|
IRF5 |
Various |
Degradation of isoforms V1 and V5, but not V2 or V3 |
[107] |
||
|
IRF7 |
K48 |
Degradation |
[108] |
||
|
IRF8 |
NA* |
Activation |
[109] |
||
|
TRIM22 |
RNF94 STAF50 |
HIV Gag, LTR |
Degradation |
[110] |
|
|
Influenza A Virus NP |
Degradation |
[111] |
|||
|
HCV NS5A |
K48 (?) |
Degradation |
[112] |
||
|
TAB2 |
K48 (?) |
Degradation |
[113] |
||
|
TRIM23 |
RNF46 ARD1 ARFD1 |
TRAF3 |
Interaction |
Function not clear, likely promoting TRAF3-mediated antiviral activity |
[114] |
|
TRAF6 |
Interaction |
Activation of NFκB mediated by HCMV UL144 |
[115] |
||
|
NEMO |
K27 |
Activation |
[114] |
||
|
TBK1 |
K27 of TRIM23 (self) |
Recruits and activates TBK1, inducing TBK1-mediated autophagy |
[116] |
||
|
TRIM24 |
RNF82 TIF1A |
TRAF3 |
K63 |
Activation |
[117] |
|
RARα |
Interaction |
Inhibits RARα activity and retinoic acid-induced STAT1 expression |
[118] |
||
|
p53 |
K48 (?) |
Promotes p53 ubiquitination and degradation |
[119] |
||
|
TRIM25 |
RNF147 ZNF147 |
Influenza virus vRNP |
Blocks vRNA chain elongation |
[120] |
|
|
RIG-I |
K63 |
Activation |
|||
|
MAVS |
K48 |
Degradation |
[123] |
||
|
ISG15 |
Functions as an ISG15 E3 ligase |
[124] |
|||
|
ZAP |
K48, K63 |
Critical for ZAP inhibition of viral genome translation |
[125] |
||
|
TRIM26 |
RNF95 ZNF173 AFP |
TBK1 |
K27 of TRIM26 (self) |
Bridges TBK1-NEMO interaction, leading to TBK1 activation |
[126] |
|
IRF3 |
K48 |
Degradation |
[127] |
||
|
TRIM27 |
RNF76 RFP |
TBK1 |
K48 |
Degradation |
|
|
IKKα, IKKβ, IKKε |
Interaction |
Inhibition |
[131] |
||
|
TRIM28 |
RNF96 KAP1 |
IRF7 |
Sumoylation |
Inhibition |
[132] |
|
TRIM29 |
ATDC |
STING |
K48 |
Degradation |
|
|
MAVS |
K11 |
Degradation |
[135] |
||
|
NEMO |
K48 |
Degradation |
[136] |
||
|
TRIM30α |
RPT1 |
STING |
K48 |
Degradation |
[137] |
|
TAB2/3 |
Lysosomal degradation |
[138] |
|||
|
TRIM31 |
RNF HCG1 |
MAVS |
K63 |
Promotes MAVS signalosome assembly |
[139] |
|
TRIM32 |
TATIP BBS11 HT2A |
Influenza PB1 |
K48 |
Degradation |
[140] |
|
STING |
K63 |
Activation |
[141] |
||
|
TRIF |
NA* |
Targets TRIF for TAX1BP1-mediated autophagic degradation |
[142] |
||
|
TRIM33 |
TIF1γ |
HIV integrase |
K48 |
Degradation |
[143] |
|
TRIM35 |
HLS5 MAIR |
TRAF3 |
K63 |
Activation |
[144] |
|
IRF7 |
K48 |
Degradation |
[145] |
||
|
TRIM38 |
RNF15 RORET |
RIG-I, MDA5 |
Sumoylation |
Stabilization |
[146] |
|
cGAS, STING |
Sumoylation |
Stabilization |
[147] |
||
|
TRAF6 |
K48 |
Degradation |
[148] |
||
|
NAP1 |
K48 |
Degradation |
[149] |
||
|
TAB2 |
K48? |
Degradation |
[150] |
||
|
TRIF |
K48 |
Degradation |
|||
|
TRIM39 |
RNF23 TFP |
Cactin |
NA* |
Stabilizes Cactin, inhibiting NFκB and IRFs |
[152] |
|
TRIM40 |
RNF35 |
RIG-I, MDA5 |
K27, K48 |
Degradation |
[153] |
|
NEMO |
Neddylation |
Inhibition |
[154] |
||
|
TRIM41 |
RINCK MGC1127 |
cGAS |
Mono-Ub |
Activation |
[155] |
|
TRIM44 |
DIPB AN3 |
MAVS |
Interaction |
Stabilization of MAVS by preventing its ubiquitination |
[156] |
|
TRIM45 |
RNF99 |
NFκB |
E3 ligase activity not required |
Inhibition of TNFα-mediated NFκB activation |
[157] |
|
TRIM56 |
RNF109 |
Influenza virus RNA |
Inhibits vRNA synthesis |
[158] |
|
|
cGAS |
Mono-Ub |
Activation |
[159] |
||
|
STING |
K63 |
Activation |
[160] |
||
|
TRIF |
Interaction |
Activation |
[161] |
||
|
TRIM59 |
RNF104 TSBF1 MRF1 IFT80L |
ECSIT |
Interaction |
Inhibition of TLR singling pathways to activate NFκB and IRFs |
[162] |
|
TRIM62 |
DEAR1 |
TRIF |
NA* |
Activation |
[86] |
|
TRIM65 |
MDA5 |
K63 |
Activation |
||
|
TRIM68 |
RNF137 SS56 |
TFG |
various |
Induces TFG lysosomal degradation |
[163] |
* NA: not assayed. Question marks (?) refer to “very likely but not experimentally revealed”.
4. TRIMs in Regulating PRR Signaling Pathways to IFN-I Production
Upon binding to PAMPs or DAMPs, PRRs trigger signals that transmit via unique adaptors to the Ub E3 ligase TRAF6 or -3 and then orchestrate to activate the kinase cascades IKKs, IRAKs, and MAPKs for the activation of the transcription factors NFκB, IRFs, and AP1. Ubiquitination regulates the cellular trafficking, stability, complex assembly, and activity of different components in PRR signaling cascades (Figure 3).
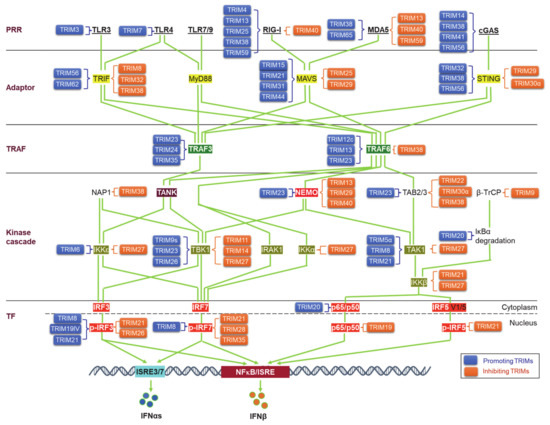
Figure 3. TRIM regulation of pathogen recognition receptor (PRR) pathways. TRIMs are involved in the regulation of the stability and activity of PRR components in all cascades of the signaling pathways, including ligands, receptors, adaptors, TRAFs, kinases and associated regulators, and the final transcription factors (TFs). An increasing pool of regulatory factors of the PRR pathways is also regulated by TRIMs (not shown). TRIMs promoting the stability or activity of the targets are shown on the left of the targets (blue), and those inhibiting the targets are shown on the right of the targets (brown). TRIM19IV and TRIM21 positively regulate phosphorylated IRF3 in indirect manners via Pin1. As such, TRIM8 positively regulates phosphorylated IRF7 in an indirect manner via Pin1. Other indirect regulations of these PRR pathways by TRIMs are not shown. TF: Transcription factor.
Viral PAMPs and other viral components can be targeted by the host Ub system, including a subset of TRIMs, for ubiquitination-mediated degradation in most cases, and thus the IFN-I response is blocked at the very beginning to suppress viral replication, with some examples listed in Table 1 [54][58]. Of note, TRIM5α (also TRIM22) targets HIV1 Gag and plays a unique role in restricting HIV1 infection (and other retroviruses), implicating a potential clinical application [61][62]. In fewer cases, TRIMs can promote viral entry and replication by targeting viral proteins. For example, TRIM7 targets Zika virus envelope protein E for K63-linked ubiquitination that enhances viral attachment to the cell surface and promotes viral entry [66]. VP35, the Ebola virus polymerase co-factor, has IFN-I inhibitory activity. TRIM6 promotes VP35 polyubiquitination to enhance viral infection [64].
5. TRIMs in Regulating the Jak-STAT IFN-I Signaling
The level of initial IFN-Is produced downstream of PRR pathways upon viral infection is relatively low due to the low level of endogenous IRF7 protein; these priming IFN-Is then secret to outside of the cell in autocrine and paracrine manners, and bind to IFN-I receptor (IFNAR) on other cells, consequently triggering the Jak-STAT IFN-I pathway, which serves as the second phase of antiviral response by inducing the expression of more IRF7, which in turn participates in IFN-I production downstream of PRR signaling, therefore amplifying the IFN-I production in a positive regulatory circuit (Figure 4) [42].
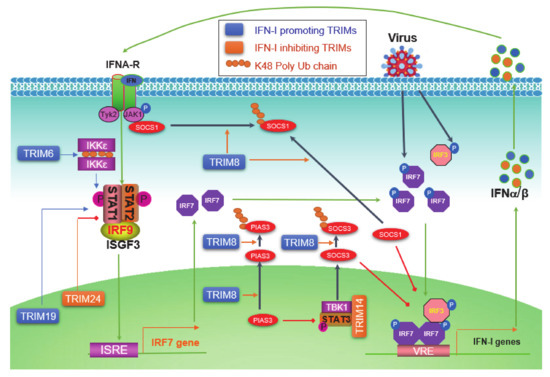
Figure 4. TRIM regulation of the Jak-STAT IFN-I signaling circuit. pathogen-associated molecular patterns (PAMPs) from pathogens activate PRR pathways, leading to the phosphorylation of constitutive high level of IRF3 and low level of IRF7, which induce a low level of IFN-Is. IFN-Is secrete from the cell and bind to IFN-I receptors (IFNAR1 and -2) on the cell membrane, followed by the recruitment and activation of Jak1 and Tyk2, leading to the phosphorylation and activation of STAT1 and -2. Phosphorylated STAT1 and -2 bind to IRF9 to form the ISGF3 (IFN-stimulated gene factor) complex, which functions as the transcriptional activator of more than 300 IFN-inducible genes (ISGs), including IRF7 itself. The induced IRF7 proteins then return to be activated by PRR pathways, and therefore constitute a positive regulatory circuit between IRF7 and IFN-Is, ensuring a potent production of IFN-Is to fight the invading pathogen. TRIM6 promotes free K48-linked Ub chains that serve as a platform to facilitate IKKε dimerization and activation. TRIM8 regulates the Jak-STAT IFN-I signaling at multiple points. The nuclear TRIM19 potentiates the transcription and activation of STAT1 and -2, and nuclear TRIM24 inhibits RARα-mediated STAT1 promoter activation. ISRE: Interferon-stimulated response element; VRE: Virus-responsive element.
Jak-STAT pathways are well known to be negatively regulated by two families: SOCS (Suppressor of cytokine signaling) and PIAS (Protein inhibitor of activated STAT). TRIM8 can shuttle between the cytoplasm and the nucleus [74], and has multiple functions to promote IFN-I signaling. TRIM8 promotes proteasomal degradation of SOCS1 and PIAS3 presumedly in the cytoplasm, and also nuclear TRIM8 promotes PIAS3 nucleus-cytoplasm translocation to inhibit PIAS3 activity [72][73][74]. SOCS1 not only inhibits Jak1 activity by directly binding to phosphorylated Jak1 in the IFN-I Jak-STAT signaling but also acts as a ubiquitin E3 ligase that targets phosphorylated IRF3 and IRF7 (both also targeted by SOCS3 that recruits the Cul-RBX2 E3 complex) for proteasomal degradation in the nucleus [164]. As such, PIAS3 acts as a SUMO E3 ligase that inhibits IRF1 transcriptional activity through sumoylation in addition to its ability to inhibit STAT3 [165]. TRIM14 negatively regulates IFN-I signaling in mouse macrophage in response to Mycobacterium tuberculosis infection by serving as a scaffold that bridges TBK1-STAT3 interaction promoting STAT3 S727 phosphorylation, consequently inducing SOCS3 expression that inhibits IFN-I signaling by targeting phosphorylated IRF3 and IRF7 as well as TBK1 for proteasomal degradation [83]. The nuclear protein TRIM19/PML promotes ISGF3-mediated gene expression by facilitating STAT1 gene transcription and STAT2 protein stabilization, as well as the accumulation of both activated STAT1 and -2 to chromosome [90].
IKKε is not only responsible for the activation of IRF7 and -3 but also plays a role in balancing IFN-I and IFN-II Jak-STAT signaling pathways in immune responses [166]. TRIM6 catalyzes free chains of K48, which promotes IKKε oligomerization and activation to facilitate STAT1 S708 phosphorylation and IFN-I signaling [65]. However, TRIM24 can inhibit retinoic acid-induced STAT1 transcription by interacting with the transcription factor RARα on the STAT1 gene promoter [118].
IFN-Is establish an antiviral state in both virus-infected cells and uninfected bystander cells, by inducing the expression of over 300 ISGs (IFN-stimulated genes) [6]. Many components of the PRR signaling pathways, such as RIG-I, cGAS, STING, IRF1, and IRF7 belong to ISGs. In addition to these components, many other ISGs, including some TRIMs themselves, are also directly regulated by TRIMs. For example, TRIM11 promotes TRIM5 turnover dependently on its RING domain [78]. Ubiquitination-like modifications, such as sumoylation and ISGylation, are involved in IFN-I-mediated defense mechanisms [30][34][167][168][169][170]. TRIM19/PML mediates global sumoylation [92], and TRIM25 functions as an ISG15 E3 ligase that mediates ISGylation [124]. Further, TRIM25 has been recently reported to be required for the stability of several ISG products [171]. The zinc-finger antiviral protein ZAP, as an ISG, is activated by TRIM25-mediated ubiquitination to inhibit viral genome translation [125]. The tumor suppressor p53 is also an ISG inducible by IFN-Is [172]. TRIM24 promotes p53 ubiquitination and degradation and, in turn, is inducible by p53 [119]. ATM phosphorylates TRIM24 at S768 and promotes its degradation, stabilizing p53 [173]. Numerous TRIMs, in addition to TRIM24, regulate p53 activity and stability in direct or indirect manners [174].
This entry is adapted from the peer-reviewed paper 10.3390/v13020279
References
- Li, S.; Gong, M.; Zhao, F.; Shao, J.; Xie, Y.; Zhang, Y.; Chang, H. Type I Interferons: Distinct Biological Activities and Current Applications for Viral Infection. Cell. Physiol. Biochem. 2018, 51, 2377–2396.
- Sandler, N.G.; Bosinger, S.E.; Estes, J.D.; Zhu, R.T.R.; Tharp, G.K.; Boritz, E.; Levin, D.; Wijeyesinghe, S.; Makamdop, K.N.; del Prete, G.Q.; et al. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature 2014, 511, 601–605.
- Mogensen, T.; Melchjorsen, J.; Larsen, C.; Paludan, S. Innate immune recognition and activation during HIV infection. Retrovirology 2010, 7, 54.
- Cha, L.; Berry, C.M.; Nolan, D.; Castley, A.; Fernandez, S.; French, M.A. Interferon-alpha, immune activation and immune dysfunction in treated HIV infection. Clin. Trans. Immunol. 2014, 3, e10.
- Crouse, J.; Kalinke, U.; Oxenius, A. Regulation of antiviral T cell responses by type I interferons. Nat. Rev. Immunol. 2015, 15, 231–242.
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103.
- Catalfamo, M.; Wilhelm, C.; Tcheung, L.; Proschan, M.; Friesen, T.; Park, J.-H.; Adelsberger, J.; Baseler, M.; Maldarelli, F.; Davey, R.; et al. CD4 and CD8 T Cell Immune Activation during Chronic HIV Infection: Roles of Homeostasis, HIV, Type I IFN, and IL-7. J. Immunol. 2011, 186, 2106–2116.
- Tough, D.F. Modulation of T-cell function by type I interferon. Immunol. Cell Biol. 2012, 90, 492–497.
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414.
- Gajewski, T.F.; Corrales, L. New perspectives on type I IFNs in cancer. Cytokine Growth Factor Rev. 2015, 26, 175–178.
- Dominguez-Villar, M.; Gautron, A.S.; de Marcken, M.; Keller, M.J.; Hafler, D.A. TLR7 induces anergy in human CD4+ T cells. Nat. Immunol. 2015, 16, 118–128.
- Wilson, E.B.; Yamada, D.H.; Elsaesser, H.; Herskovitz, J.; Deng, J.; Cheng, G.; Aronow, B.J.; Karp, C.L.; Brooks, D.G. Blockade of chronic Type I interferon signaling to control persistent LCMV infection. Science 2013, 340, 202–207.
- Teijaro, J.R.; Ng, C.; Lee, A.M.; Sullivan, B.M.; Sheehan, K.C.F.; Welch, M.; Schreiber, R.D.; Carlos de la Torre, J.; Oldstone, M.B.A. Persistent LCMV infection is controlled by blockade of Type I interferon signaling. Science 2013, 340, 207–211.
- Bosque, A.; Planelles, V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 2009, 113, 58–65.
- Gonzalez-Navajas, J.M.; Lee, J.; David, M.; Raz, E. Immunomodulatory functions of type I interferons. Nat. Rev. Immunol. 2012, 12, 125–135.
- Bonjardim, C.A.; Ferreira, P.C.P.; Kroon, E.G. Interferons: Signaling, antiviral and viral evasion. Immunol. Lett. 2009, 122, 1–11.
- Hervas-Stubbs, S.; Perez-Gracia, J.L.; Rouzaut, A.; Sanmamed, M.F.; Le Bon, A.; Melero, I. Direct Effects of Type I Interferons on Cells of the Immune System. Clin. Cancer Res. 2011, 17, 2619–2627.
- Forster, S. Interferon signatures in immune disorders and disease. Immunol. Cell Biol. 2012, 90, 520–527.
- Elkon, K.B.; Wiedeman, A. Type I IFN system in the development and manifestations of SLE. Curr.Opin. Rheumatol. 2012, 24, 499–505.
- von Locquenghien, M.; Rozalén, C.; Celià-Terrassa, T. Interferons in cancer immunoediting: Sculpting metastasis and immunotherapy response. J. Clin. Investig. 2021, 131.
- Arimoto, K.-I.; Miyauchi, S.; Stoner, S.A.; Fan, J.-B.; Zhang, D.-E. Negative regulation of type I IFN signaling. J. Leukoc. Biol. 2018, 103, 1099–1116.
- Wang, L.; Ning, S. “Toll-free” pathways for production of type I interferons. Allergy Immunol. 2017, 1, 143–163.
- McClellan, A.J.; Laugesen, S.H.; Ellgaard, L. Cellular functions and molecular mechanisms of non-lysine ubiquitination. Open Biol. 2019, 9, 190147.
- Huang, Q.; Zhang, X. Emerging Roles and Research Tools of Atypical Ubiquitination. Proteomics 2020, 20, 1900100.
- Hrdinka, M.; Gyrd-Hansen, M. The Met1-Linked Ubiquitin Machinery: Emerging Themes of (De)regulation. Mol. Cell 2017, 68, 265–280.
- Iwai, K. Discovery of linear ubiquitination, a crucial regulator for immune signaling and cell death. FEBS J. 2020.
- Oikawa, D.; Sato, Y.; Ito, H.; Tokunaga, F. Linear Ubiquitin Code: Its Writer, Erasers, Decoders, Inhibitors, and Implications in Disorders. Int. J. Mol. Sci. 2020, 21, 3381.
- Ferrarelli, L.K. Palmitoylation makes the switch for EGFR. Science 2020, 367, 1086–1088.
- Wei, Y.; Xu, X. UFMylation: A Unique & Fashionable Modification for Life. Genom. Proteom. Bioinform. 2016, 14, 140–146.
- Zhang, Y.; Wang, Y.; Zhu, C.; Robertson, E.S.; Cai, Q. Role of SUMOylation in Human Oncogenic Herpesvirus Infection. Virus Res. 2020.
- Su, S.; Zhang, Y.; Liu, P. Roles of Ubiquitination and SUMOylation in DNA Damage Response. Curr. Issues Mol. Biol. 2019, 35, 59–84.
- Yu, J.; Qin, B.; Lou, Z. Ubiquitin and ubiquitin-like molecules in DNA double strand break repair. Cell Biosci. 2020, 10, 13.
- Chang, S.C.; Ding, J.L. Ubiquitination and SUMOylation in the chronic inflammatory tumor microenvironment. Biochim. Biophys. Acta BBA Rev. Cancer 2018, 1870, 165–175.
- Perng, Y.-C.; Lenschow, D.J. ISG15 in antiviral immunity and beyond. Nat. Rev. Microbiol. 2018, 16, 423–439.
- Fiil, B.K.; Gyrd-Hansen, M. The Met1-linked ubiquitin machinery in inflammation and infection. Cell Death Differ. 2021, 28, 557–569.
- Masucci, M.G. Epstein-Barr virus oncogenesis and the ubiquitin-proteasome system. Oncogene 2004, 23, 2107–2115.
- Đukić, A.; Lulić, L.; Thomas, M.; Skelin, J.; Bennett Saidu, N.E.; Grce, M.; Banks, L.; Tomaić, V. HPV Oncoproteins and the Ubiquitin Proteasome System: A Signature of Malignancy? Pathogens 2020, 9, 133.
- Shackelford, J.; Pagano, J. Role of the ubiquitin system and tumor viruses in AIDS-related cancer. BMC Biochem. 2007, 8, S8.
- Zheng, Y.; Gao, C. Chapter Four—Fine-tuning of antiviral innate immunity by ubiquitination. Adv. Immunol. 2020, 145, 95–128.
- Ashizawa, A.; Higashi, C.; Masuda, K.; Ohga, R.; Taira, T.; Fujimuro, M. The Ubiquitin System and Kaposi’s Sarcoma-Associated Herpesvirus. Front. Microbiol. 2012, 3, 66.
- Lopez-Castejon, G. Control of the inflammasome by the ubiquitin system. FEBS J. 2020, 287, 11–26.
- Ning, S.; Pagano, J.; Barber, G. IRF7: Activation, regulation, modification, and function. Genes Immun. 2011, 12, 399–414.
- Härtlova, A.; Erttmann, S.F.; Raffi, F.A.M.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kröger, A.; Nilsson, J.A.; et al. DNA Damage Primes the Type I Interferon System via the Cytosolic DNA Sensor STING to Promote Anti-Microbial Innate Immunity. Immunity 2015, 42, 332–343.
- Schlee, M.; Hartmann, G. Discriminating self from non-self in nucleic acid sensing. Nat. Rev. Immunol. 2016, 16, 566–580.
- White, M.J.; McArthur, K.; Metcalf, D.; Lane, R.M.; Cambier, J.C.; Herold, M.J.; van Delft, M.F.; Bedoui, S.; Lessene, G.; Ritchie, M.E.; et al. Apoptotic Caspases Suppress mtDNA-Induced STING-Mediated Type I IFN Production. Cell 2014, 159, 1549–1562.
- de Galarreta, M.R.; Lujambio, A. DNA sensing in senescence. Nat. Cell Biol. 2017, 19, 1008–1009.
- Gluck, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070.
- Agalioti, T.; Lomvardas, S.; Parekh, B.; Yie, J.; Maniatis, T.; Thanos, D. Ordered recruitment of chromatin modifying and general transcription factors to the IFN-α promoter. Cell 2000, 103, 667–678.
- Hatakeyama, S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311.
- van Tol, S.; Hage, A.; Giraldo, M.; Bharaj, P.; Rajsbaum, R. The TRIMendous Role of TRIMs in Virus–Host Interactions. Vaccines 2017, 5, 23.
- Sardiello, M.; Cairo, S.; Fontanella, B.; Ballabio, A.; Meroni, G. Genomic analysis of the TRIM family reveals two groups of genes with distinct evolutionary properties. BMC Evol. Biol. 2008, 8, 225.
- Versteeg, G.A.; Benke, S.; García-Sastre, A.; Rajsbaum, R. InTRIMsic immunity: Positive and negative regulation of immune signaling by tripartite motif proteins. Cytokine Growth Factor Rev. 2014, 25, 563–576.
- Yang, W.; Gu, Z.; Zhang, H.; Hu, H. To TRIM the Immunity: From Innate to Adaptive Immunity. Front. Immunol. 2020, 11.
- Giraldo, M.I.; Hage, A.; van Tol, S.; Rajsbaum, R. TRIM Proteins in Host Defense and Viral Pathogenesis. Curr. Clin. Microbiol. Rep. 2020, 7, 101–114.
- Patil, G.; Li, S. Tripartite motif proteins: An emerging antiviral protein family. Future Virol. 2019, 14, 107–122.
- Di Rienzo, M.; Romagnoli, A.; Antonioli, M.; Piacentini, M.; Fimia, G.M. TRIM proteins in autophagy: Selective sensors in cell damage and innate immune responses. Cell Death Differ. 2020, 27, 887–902.
- Khan, R.; Khan, A.; Ali, A.; Idrees, M. The interplay between viruses and TRIM family proteins. Rev. Med. Virol. 2019, 29, e2028.
- Hage, A.; Rajsbaum, R. To TRIM or not to TRIM: The balance of host-virus interactions mediated by the ubiquitin system. J. Gen. Virol. 2019, 100, 1641–1662.
- Li, W.-W.; Nie, Y.; Yang, Y.; Ran, Y.; Luo, W.-W.; Xiong, M.-G.; Wang, S.-Y.; Xu, Z.-S.; Wang, Y.-Y. Ubiquitination of TLR3 by TRIM3 signals its ESCRT-mediated trafficking to the endolysosomes for innate antiviral response. Proc. Natl. Acad. Sci. USA 2020, 117, 23707–23716.
- Yan, J.; Li, Q.; Mao, A.P.; Hu, M.M.; Shu, H.B. TRIM4 modulates type I interferon induction and cellular antiviral response by targeting RIG-I for K63-linked ubiquitination. J. Mol. Cell Biol. 2014, 6, 154–163.
- Ganser-Pornillos, B.K.; Pornillos, O. Restriction of HIV-1 and other retroviruses by TRIM5. Nat. Rev. Microbiol. 2019, 17, 546–556.
- Chang, T.-H.; Yoshimi, R.; Ozato, K. TRIM12c, a Mouse Homolog of TRIM5, Is a Ubiquitin Ligase That Stimulates Type I IFN and NF-κB Pathways along with TNFR-Associated Factor 6. J. Immunol. 2015, 195, 5367–5379.
- Pertel, T.; Hausmann, S.; Morger, D.; Zuger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365.
- Bharaj, P.; Atkins, C.; Luthra, P.; Giraldo, M.I.; Dawes, B.E.; Miorin, L.; Johnson, J.R.; Krogan, N.J.; Basler, C.F.; Freiberg, A.N.; et al. The Host E3-Ubiquitin Ligase TRIM6 Ubiquitinates the Ebola Virus VP35 Protein and Promotes Virus Replication. J. Virol. 2017, 91, e00833-17.
- Rajsbaum, R.; Versteeg, G.A.; Schmid, S.; Maestre, A.M.; Belicha-Villanueva, A.; Martínez-Romero, C.; Patel, J.R.; Morrison, J.; Pisanelli, G.; Miorin, L.; et al. Unanchored K48-linked polyubiquitin synthesized by the E3-ubiquitin ligase TRIM6 stimulates the interferon-IKKε kinase-mediated antiviral response. Immunity 2014, 40, 880–895.
- Giraldo, M.I.; Xia, H.; Aguilera-Aguirre, L.; Hage, A.; van Tol, S.; Shan, C.; Xie, X.; Sturdevant, G.L.; Robertson, S.J.; McNally, K.L.; et al. Envelope protein ubiquitination drives entry and pathogenesis of Zika virus. Nature 2020, 585, 414–419.
- Lu, M.; Zhu, X.; Yang, Z.; Zhang, W.; Sun, Z.; Ji, Q.; Chen, X.; Zhu, J.; Wang, C.; Nie, S. E3 ubiquitin ligase TRIM7 positively regulates the TLR4-mediated immune response via its E3 ligase domain in macrophages. Mol. Immunol. 2019, 109, 126–133.
- Ye, W.; Hu, M.-M.; Lei, C.-Q.; Zhou, Q.; Lin, H.; Sun, M.-S.; Shu, H.-B. TRIM8 Negatively Regulates TLR3/4-Mediated Innate Immune Response by Blocking TRIF–TBK1 Interaction. J. Immunol. 2017, 199, 1856–1864.
- Li, Q.; Yan, J.; Mao, A.P.; Li, C.; Ran, Y.; Shu, H.B.; Wang, Y.Y. TRIM8 modulates TNFà- and IL-1á-triggered NF- k B activation by targeting TAK1 for K63-linked polyubiquitination. Proc. Natl. Acad. Sci. USA 2011, 108, 19341–19346.
- Guo, L.; Dong, W.; Fu, X.; Lin, J.; Dong, Z.; Tan, X.; Zhang, T. Tripartite Motif 8 (TRIM8) Positively Regulates Pro-inflammatory Responses in Pseudomonas aeruginosa-Induced Keratitis Through Promoting K63-Linked Polyubiquitination of TAK1 Protein. Inflammation 2017, 40, 454–463.
- Maarifi, G.; Smith, N.; Maillet, S.; Moncorgé, O.; Chamontin, C.; Edouard, J.; Sohm, F.; Blanchet, F.P.; Herbeuval, J.-P.; Lutfalla, G.; et al. TRIM8 is required for virus-induced IFN response in human plasmacytoid dendritic cells. Sci. Adv. 2019, 5, eaax3511.
- Toniato, E.; Chen, X.P.; Losman, J.; Flati, V.; Donahue, L.; Rothman, P. TRIM8/GERP RING finger protein interacts with SOCS-1. J. Biol. Chem. 2002, 277, 37315–37322.
- Okumura, F.; Matsunaga, Y.; Katayama, Y.; Nakayama, K.I.; Hatakeyama, S. TRIM8 modulates STAT3 activity through negative regulation of PIAS3. J. Cell Sci. 2010, 123, 2238–2245.
- Tomar, D.; Sripada, L.; Prajapati, P.; Singh, R.; Singh, A.K.; Singh, R. Nucleo-Cytoplasmic Trafficking of TRIM8, a Novel Oncogene, Is Involved in Positive Regulation of TNF Induced NF-κB Pathway. PLoS ONE 2012, 7, e48662.
- Qin, Y.; Liu, Q.; Tian, S.; Xie, W.; Cui, J.; Wang, R.F. TRIM9 short isoform preferentially promotes DNA and RNA virus-induced production of type I interferon by recruiting GSK3β to TBK1. Cell Res. 2016, 26, 613–628.
- Shi, M.; Cho, H.; Inn, K.-S.; Yang, A.; Zhao, Z.; Liang, Q.; Versteeg, G.A.; Amini-Bavil-Olyaee, S.; Wong, L.-Y.; Zlokovic, B.V.; et al. Negative regulation of NF-κB activity by brain-specific TRIpartite Motif protein 9. Nat. Commun. 2014, 5, 4820.
- Lee, Y.; Song, B.; Park, C.; Kwon, K.S. TRIM11 negatively regulates IFNβ production and antiviral activity by targeting TBK1. PLoS ONE 2013, 8, e63255.
- Uchil, P.D.; Quinlan, B.D.; Chan, W.T.; Luna, J.M.; Mothes, W. TRIM E3 ligases interfere with early and late stages of the retroviral life cycle. PLoS Pathog. 2008, 4, e16.
- Narayan, K.; Waggoner, L.; Pham, S.T.; Hendricks, G.L.; Waggoner, S.N.; Conlon, J.; Wang, J.P.; Fitzgerald, K.A.; Kang, J. TRIM13 Is a Negative Regulator of MDA5-Mediated Type I Interferon Production. J. Virol. 2014, 88, 10748–10757.
- Huang, B.; Baek, S.H. Trim13 Potentiates Toll-Like Receptor 2-Mediated Nuclear Factor κB Activation via K29-Linked Polyubiquitination of Tumor Necrosis Factor Receptor-Associated Factor 6. Mol. Pharm. 2017, 91, 307–316.
- Tomar, D.; Singh, R. TRIM13 regulates ubiquitination and turnover of NEMO to suppress TNF induced NF-κB activation. Cell. Signal. 2014, 26, 2606–2613.
- Wang, S.; Chen, Y.; Li, C.; Wu, Y.; Guo, L.; Peng, C.; Huang, Y.; Cheng, G.; Qin, F. TRIM14 inhibits hepatitis C virus infection by SPRY domain-dependent targeted degradation of the viral NS5A protein. Sci. Rep. 2016, 6, 32336.
- Hoffpauir, C.T.; Bell, S.L.; West, K.O.; Jing, T.; Wagner, A.R.; Torres-Odio, S.; Cox, J.S.; West, A.P.; Li, P.; Patrick, K.L.; et al. TRIM14 Is a Key Regulator of the Type I IFN Response during Mycobacterium tuberculosis Infection. J. Immunol. 2020, 205, 153–167.
- Zhou, Z.; Jia, X.; Xue, Q.; Dou, Z.; Ma, Y.; Zhao, Z.; Jiang, Z.; He, B.; Jin, Q.; Wang, J. TRIM14 is a mitochondrial adaptor that facilitates RIG-I–like receptor-mediated innate immune response. Proc. Natl. Acad. Sci. USA 2014, 111, E245–E254.
- Chen, M.; Meng, Q.; Qin, Y.; Liang, P.; Tan, P.; He, L.; Zhou, Y.; Chen, Y.; Huang, J.; Wang, R.-F.; et al. TRIM14 Inhibits cGAS Degradation Mediated by Selective Autophagy Receptor p62 to Promote Innate Immune Responses. Mol. Cell 2016, 64, 105–119.
- Uchil, P.D.; Hinz, A.; Siegel, S.; Coenen-Stass, A.; Pertel, T.; Luban, J.; Mothes, W. TRIM Protein-Mediated Regulation of Inflammatory and Innate Immune Signaling and Its Association with Antiretroviral Activity. J. Virol. 2013, 87, 257–272.
- Turelli, P.; Doucas, V.; Craig, E.; Mangeat, B.; Klages, N.; Evans, R.; Kalpana, G.; Trono, D. Cytoplasmic recruitment of INI1 and PML on incoming HIV preintegration complexes: Interference with early steps of viral replication. Mol. Cell 2001, 7, 1245–1254.
- Regad, T.; Saib, A.; Lallemand-Breitenbach, V.; Pandolfi, P.P.; de Thé, H.; Chelbi-Alix, M.K. PML mediates the interferon-induced antiviral state against a complex retrovirus via its association with the viral transactivator. EMBO J. 2001, 20, 3495–3505.
- Djavani, M.; Rodas, J.; Lukashevich, I.S.; Horejsh, D.; Pandolfi, P.P.; Borden, K.L.B.; Salvato, M.S. Role of the Promyelocytic Leukemia Protein PML in the Interferon Sensitivity of Lymphocytic Choriomeningitis Virus. J. Virol. 2001, 75, 6204–6208.
- Kim, Y.-E.; Ahn, J.-H. Positive Role of PML Protein in Type I Interferon Response and Its Regulation by Human Cytomegalovirus. PLoS Pathog. 2015, 11, e1004785.
- El Asmi, F.; Maroui, M.A.; Dutrieux, J.; Blondel, D.; Nisole, S.; Chelbi-Alix, M.K. Implication of PMLIV in both intrinsic and innate immunity. PLoS Pathog. 2014, 10, e1003975.
- Maroui, M.A.; Maarifi, G.; McManus, F.P.; Lamoliatte, F.; Thibault, P.; Chelbi-Alix, M.K. Promyelocytic Leukemia Protein (PML) Requirement for Interferon-induced Global Cellular SUMOylation. Mol. Cell. Proteom. 2018, 17, 1196–1208.
- Wu, W.-S.; Xu, Z.-X.; Hittelman, W.N.; Salomoni, P.; Pandolfi, P.P.; Chang, K.-S. Promyelocytic Leukemia Protein Sensitizes Tumor Necrosis Factor alpha-Induced Apoptosis by Inhibiting the NF-kB Survival Pathway. J. Biol. Chem. 2003, 278, 12294–12304.
- Ahmed, A.; Wan, X.; Mitxitorena, I.; Lindsay, A.J.; Paolo Pandolfi, P.; McCaffrey, M.W.; Keeshan, K.; Chen, Y.H.; Carmody, R.J. Regulation of NF-κB by PML and PML-RARα. Sci. Rep. 2017, 7, 44539.
- Niwa-Kawakita, M.; Ferhi, O.; Soilihi, H.; Le Bras, M.; Lallemand-Breitenbach, V.; de Thé, H. PML is a ROS sensor activating p53 upon oxidative stress. J. Exp. Med. 2017, 214, 3197–3206.
- Chae, J.J.; Wood, G.; Richard, K.; Jaffe, H.; Colburn, N.T.; Masters, S.L.; Gumucio, D.L.; Shoham, N.G.; Kastner, D.L. The familial Mediterranean fever protein, pyrin, is cleaved by caspase-1 and activates NF-kappaB through its N-terminal fragment. Blood 2008, 112, 1794–1803.
- Zhang, Z.; Bao, M.; Lu, N.; Weng, L.; Yuan, B.; Liu, Y.J. The E3 ubiquitin ligase TRIM21 negatively regulates the innate immune response to intracellular double-stranded DNA. Nat. Immunol. 2013, 14, 172–178.
- Xue, B.; Li, H.; Guo, M.; Wang, J.; Xu, Y.; Zou, X.; Deng, R.; Li, G.; Zhu, H. TRIM21 Promotes Innate Immune Response to RNA Viral Infection through Lys27-Linked Polyubiquitination of MAVS. J. Virol. 2018, 92.
- Young, J.A.; Sermwittayawong, D.; Kim, H.J.; Nandu, S.; An, N.; Erdjument-Bromage, H.; Tempst, P.; Coscoy, L.; Winoto, A. Fas-associated death domain (FADD) and the E3 ubiquitin-protein ligase TRIM21 interact to negatively regulate virus-induced interferon production. J. Biol. Chem. 2011, 286, 6521–6531.
- McEwan, W.A.; Tam, J.C.H.; Watkinson, R.E.; Bidgood, S.R.; Mallery, D.L.; James, L.C. Intracellular antibody-bound pathogens stimulate immune signaling via the Fc receptor TRIM21. Nat. Immunol. 2013, 14, 327–336.
- Geijtenbeek, T.B.; Gringhuis, S.I. An inside job for antibodies: Tagging pathogens for intracellular sensing. Nat. Immunol. 2013, 14, 309–311.
- Wada, K.; Niida, M.; Tanaka, M.; Kamitani, T. Ro52-mediated monoubiquitination of IKKβ down-regulates NF-κB signalling. J Biochem. 2009, 146, 821–832.
- Yang, K.; Shi, H.X.; Liu, X.Y.; Shan, Y.F.; Wei, B.; Chen, S.; Wang, C. TRIM21 is essential to sustain IFN Regulatory Factor 3 activation during antiviral response. J. Immunol. 2009, 182, 3782–3792.
- Stacey, K.B.; Breen, E.; Jefferies, C.A. Tyrosine phosphorylation of the E3 ubiquitin ligase TRIM21 positively regulates interaction with IRF3 and hence TRIM21 activity. PLoS ONE 2012, 7, e34041.
- Higgs, R.; Gabhann, J.N.; Larbi, N.B.; Breen, E.P.; Fitzgerald, K.A.; Jefferies, C.A. The E3 ubiquitin ligase Ro52 negatively regulates IFN-á production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. J. Immunol. 2008, 181, 1780–1786.
- Kimura, T.; Jain, A.; Choi, S.W.; Mandell, M.A.; Johansen, T.; Deretic, V. TRIM-directed selective autophagy regulates immune activation. Autophagy 2017, 13, 989–990.
- Lazzari, E.; Korczeniewska, J.; Ni, G.J.; Smith, S.; Barnes, B.J.; Jefferies, C.A. TRIM21 Differentially Regulates the Stability of Interferon Regulatory Factor 5 (IRF5) Isoforms. PLoS ONE 2014, 9, e103609.
- Higgs, R.; Lazzari, E.; Wynne, C.; NÃ Gabhann, J.; Espinosa, A.; Wahren-Herlenius, M.; Jefferies, C.A. Self protection from anti-Viral responses--Ro52 promotes degradation of the transcription factor IRF7 downstream of the viral toll-like receptors. PLoS ONE 2010, 5, e11776.
- Kong, H.J.; Anderson, D.E.; Lee, C.H.; Jang, M.K.; Tamura, T.; Tailor, P.; Cho, H.K.; Cheong, J.; Xiong, H.; Morse, H.C., III; et al. Cutting edge: Autoantigen Ro52 is an interferon inducible E3 ligase that ubiquitinates IRF8 and enhances cytokine expression in macrophages. J. Immunol. 2007, 179, 26–30.
- Vicenzi, E.; Poli, G. The interferon-stimulated gene TRIM22: A double-edged sword in HIV-1 infection. Cytokine Growth Factor Rev. 2018, 40, 40–47.
- Di Pietro, A.; Kajaste-Rudnitski, A.; Oteiza, A.; Nicora, L.; Towers, G.J.; Mechti, N.; Vicenzi, E. TRIM22 Inhibits Influenza A Virus Infection by Targeting the Viral Nucleoprotein for Degradation. J. Virol. 2013, 87, 4523–4533.
- Yang, C.; Zhao, X.; Sun, D.; Yang, L.; Chong, C.; Pan, Y.; Chi, X.; Gao, Y.; Wang, M.; Shi, X.; et al. Interferon alpha (IFNα)-induced TRIM22 interrupts HCV replication by ubiquitinating NS5A. Cell. Mol. Immunol. 2016, 13, 94–102.
- Qiu, H.; Huang, F.; Xiao, H.; Sun, B.; Yang, R. TRIM22 inhibits the TRAF6-stimulated NF-κB pathway by targeting TAB2 for degradation. Virol. Sin. 2013, 28, 209–215.
- Arimoto, K.i.; Funami, K.; Saeki, Y.; Tanaka, K.; Okawa, K.; Takeuchi, O.; Akira, S.; Murakami, Y.; Shimotohno, K. Polyubiquitin conjugation to NEMO by triparite motif protein 23 (TRIM23) is critical in antiviral defense. Proc. Natl. Acad. Sci. USA 2010, 107, 15856–15861.
- Poole, E.; Groves, I.; MacDonald, A.; Pang, Y.; Alcami, A.; Sinclair, J. Identification of TRIM23 as a cofactor involved in the regulation of NF-kappaB by human cytomegalovirus. J. Virol. 2009, 83, 3581–3590.
- Sparrer, K.M.J.; Gableske, S.; Zurenski, M.A.; Parker, Z.M.; Full, F.; Baumgart, G.J.; Kato, J.; Pacheco-Rodriguez, G.; Liang, C.; Pornillos, O.; et al. TRIM23 mediates virus-induced autophagy via activation of TBK1. Nat. Microbiol. 2017, 2, 1543–1557.
- Zhu, Q.; Yu, T.; Gan, S.; Wang, Y.; Pei, Y.; Zhao, Q.; Pei, S.; Hao, S.; Yuan, J.; Xu, J.; et al. TRIM24 facilitates antiviral immunity through mediating K63-linked TRAF3 ubiquitination. J. Exp. Med. 2020, 217.
- Tisserand, J.; Khetchoumian, K.; Thibault, C.; Dembélé, D.; Chambon, P.; Losson, R. TRIM24 (Trim24/Tif1α) Tumor Suppressor Protein Is a Novel Negative Regulator of Interferon (IFN)/Signal Transducers and Activators of Transcription (STAT) Signaling Pathway Acting through Retinoic Acid Receptor α (Rarα) Inhibition. J. Biol. Chem. 2011, 286, 33369–33379.
- Allton, K.; Jain, A.K.; Herz, H.-M.; Tsai, W.-W.; Jung, S.Y.; Qin, J.; Bergmann, A.; Johnson, R.L.; Barton, M.C. Trim24 targets endogenous p53 for degradation. Proc. Natl. Acad. Sci. USA 2009, 106, 11612–11616.
- Meyerson, N.R.; Zhou, L.; Guo, Y.R.; Zhao, C.; Tao, Y.J.; Krug, R.M.; Sawyer, S.L. Nuclear TRIM25 Specifically Targets Influenza Virus Ribonucleoproteins to Block the Onset of RNA Chain Elongation. Cell Host Microbe 2017, 22, 627–638.e627.
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920.
- Oshiumi, H.; Matsumoto, M.; Hatakeyama, S.; Seya, T. Riplet/RNF135, a RING-finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J. Biol. Chem. 2009, 284, 807–817.
- Castanier, C.; Zemirli, N.; Portier, A.; Garcin, D.; Bidère, N.; Vazquez, A.; Arnoult, D. MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 2012, 10, 44.
- Zou, W.; Zhang, D.-E. The Interferon-inducible Ubiquitin-protein Isopeptide Ligase (E3) EFP Also Functions as an ISG15 E3 Ligase. J. Biol. Chem. 2006, 281, 3989–3994.
- Zheng, X.; Wang, X.; Tu, F.; Wang, Q.; Fan, Z.; Gao, G. TRIM25 Is Required for the Antiviral Activity of Zinc Finger Antiviral Protein. J. Virol. 2017, 91, e00088-17.
- Ran, Y.; Zhang, J.; Liu, L.L.; Pan, Z.Y.; Nie, Y.; Zhang, H.Y.; Wang, Y.Y. Autoubiquitination of TRIM26 links TBK1 to NEMO in RLR-mediated innate antiviral immune response. J. Mol. Cell Biol. 2016, 8, 31–43.
- Wang, P.; Zhao, W.; Zhao, K.; Zhang, L.; Gao, C. TRIM26 Negatively Regulates Interferon-β Production and Antiviral Response through Polyubiquitination and Degradation of Nuclear IRF3. PLoS Pathog. 2015, 11, e1004726.
- Cai, J.; Chen, H.Y.; Peng, S.J.; Meng, J.L.; Wang, Y.; Zhou, Y.; Qian, X.P.; Sun, X.Y.; Pang, X.W.; Zhang, Y.; et al. USP7-TRIM27 axis negatively modulates antiviral type I IFN signaling. FASEB J. 2018, 32, 5238–5249.
- Zheng, F.; Xu, N.; Zhang, Y. TRIM27 Promotes Hepatitis C Virus Replication by Suppressing Type I Interferon Response. Inflammation 2019, 42, 1317–1325.
- Zheng, Q.; Hou, J.; Zhou, Y.; Yang, Y.; Xie, B.; Cao, X. Siglec1 suppresses antiviral innate immune response by inducing TBK1 degradation via the ubiquitin ligase TRIM27. Cell Res. 2015, 25, 1121–1136.
- Zha, J.; Han, K.-J.; Xu, L.-G.; He, W.; Zhou, Q.; Chen, D.; Zhai, Z.; Shu, H.-B. The Ret Finger Protein Inhibits Signaling Mediated by the Noncanonical and Canonical IκB Kinase Family Members. J. Immunol. 2006, 176, 1072–1080.
- Liang, Q.; Deng, H.; Li, X.; Wu, X.; Tang, Q.; Chang, T.H.; Peng, H.; Rauscher, F.J.; Ozato, K.; Zhu, F. TRIM28 is a small ubiquitin-related modifier E3 ligase and negative regulator of IFN Regulatory Factor 7. J. Immunol. 2011, 187, 4754–4763.
- Li, Q.; Lin, L.; Tong, Y.; Liu, Y.; Mou, J.; Wang, X.; Wang, X.; Gong, Y.; Zhao, Y.; Liu, Y.; et al. TRIM29 negatively controls antiviral immune response through targeting STING for degradation. Cell Discov. 2018, 4, 13.
- Xing, J.; Zhang, A.; Zhang, H.; Wang, J.; Li, X.C.; Zeng, M.-S.; Zhang, Z. TRIM29 promotes DNA virus infections by inhibiting innate immune response. Nat. Commun. 2017, 8, 945.
- Xing, J.; Zhang, A.; Minze, L.J.; Li, X.C.; Zhang, Z. TRIM29 Negatively Regulates the Type I IFN Production in Response to RNA Virus. J. Immunol. 2018, 201, 183–192.
- Xing, J.; Weng, L.; Yuan, B.; Wang, Z.; Jia, L.; Jin, R.; Lu, H.; Li, X.C.; Liu, Y.-J.; Zhang, Z. Identification of a role for TRIM29 in the control of innate immunity in the respiratory tract. Nat. Immunol. 2016, 17, 1373–1380.
- Wang, Y.; Lian, Q.; Yang, B.; Yan, S.; Zhou, H.; He, L.; Lin, G.; Lian, Z.; Jiang, Z.; Sun, B. TRIM30α Is a Negative-Feedback Regulator of the Intracellular DNA and DNA Virus-Triggered Response by Targeting STING. PLoS Pathog. 2015, 11, e1005012.
- Shi, M.; Deng, W.; Bi, E.; Mao, K.; Ji, Y.; Lin, G.; Wu, X.; Tao, Z.; Li, Z.; Cai, X.; et al. TRIM30à negatively regulates TLR-mediated NF- k B activation by targeting TAB2 and TAB3 for degradation. Nat. Immunol. 2008, 9, 369–377.
- Liu, B.; Zhang, M.; Chu, H.; Zhang, H.; Wu, H.; Song, G.; Wang, P.; Zhao, K.; Hou, J.; Wang, X.; et al. The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat. Immunol. 2017, 18, 214–224.
- Fu, B.; Wang, L.; Ding, H.; Schwamborn, J.C.; Li, S.; Dorf, M.E. TRIM32 Senses and Restricts Influenza A Virus by Ubiquitination of PB1 Polymerase. PLoS Pathog. 2015, 11, e1004960.
- Zhang, J.; Hu, M.M.; Wang, Y.Y.; Shu, H.B. TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J. Biol. Chem. 2012, 287, 28646–28655.
- Yang, Q.; Liu, T.-T.; Lin, H.; Zhang, M.; Wei, J.; Luo, W.-W.; Hu, Y.-H.; Zhong, B.; Hu, M.-M.; Shu, H.-B. TRIM32-TAX1BP1-dependent selective autophagic degradation of TRIF negatively regulates TLR3/4-mediated innate immune responses. PLoS Pathog. 2017, 13, e1006600.
- Ali, H.; Mano, M.; Braga, L.; Naseem, A.; Marini, B.; Vu, D.M.; Collesi, C.; Meroni, G.; Lusic, M.; Giacca, M. Cellular TRIM33 restrains HIV-1 infection by targeting viral integrase for proteasomal degradation. Nat. Commun. 2019, 10, 926.
- Sun, N.; Jiang, L.; Ye, M.; Wang, Y.; Wang, G.; Wan, X.; Zhao, Y.; Wen, X.; Liang, L.; Ma, S.; et al. TRIM35 mediates protection against influenza infection by activating TRAF3 and degrading viral PB2. Protein Cell 2020, 11, 894–914.
- Wang, Y.; Yan, S.; Yang, B.; Wang, Y.; Zhou, H.; Lian, Q.; Sun, B. TRIM35 negatively regulates TLR7- and TLR9-mediated type I interferon production by targeting IRF7. FEBS Lett. 2015, 589, 1322–1330.
- Hu, M.M.; Liao, C.Y.; Yang, Q.; Xie, X.Q.; Shu, H.B. Innate immunity to RNA virus is regulated by temporal and reversible sumoylation of RIG-I and MDA5. J. Exp. Med. 2017, 214, 973–989.
- Hu, M.-M.; Yang, Q.; Xie, X.-Q.; Liao, C.-Y.; Lin, H.; Liu, T.-T.; Yin, L.; Shu, H.-B. Sumoylation Promotes the Stability of the DNA Sensor cGAS and the Adaptor STING to Regulate the Kinetics of Response to DNA Virus. Immunity 2016, 45, 555–569.
- Zhao, W.; Wang, L.; Zhang, M.; Yuan, C.; Gao, C. E3 Ubiquitin Ligase TRIM38 Negatively Regulates TLR-Mediated Immune Responses by Proteasomal Degradation of TNF Receptor-Associated Factor 6 in Macrophages. J. Immunol. 2012, 188, 2567–2574.
- Zhao, W.; Wang, L.; Zhang, M.; Wang, P.; Yuan, C.; Qi, J.; Meng, H.; Gao, C. TRIM38 negatively regulates TLR3/4- and RIG-I-mediated IFN-β production and antiviral response by targeting NAP1. J. Immunol. 2012, 188, 5311–5318.
- Hu, M.M.; Xie, X.Q.; Yang, Q.; Liao, C.Y.; Ye, W.; Lin, H.; Shu, H.B. TRIM38 Negatively Regulates TLR3/4-Mediated Innate Immune and Inflammatory Responses by Two Sequential and Distinct Mechanisms. J. Immunol. 2015, 195, 4415–4425.
- Xue, Q.; Zhou, Z.; Lei, X.; Liu, X.; He, B.; Wang, J.; Hung, T. TRIM38 Negatively Regulates TLR3-Mediated IFN-β Signaling by Targeting TRIF for Degradation. PLoS ONE 2012, 7, e46825.
- Suzuki, M.; Watanabe, M.; Nakamaru, Y.; Takagi, D.; Takahashi, H.; Fukuda, S.; Hatakeyama, S. TRIM39 negatively regulates the NFκB-mediated signaling pathway through stabilization of Cactin. Cell Mol. Life Sci. 2016, 73, 1085–1101.
- Zhao, C.; Jia, M.; Song, H.; Yu, Z.; Wang, W.; Li, Q.; Zhang, L.; Zhao, W.; Cao, X. The E3 Ubiquitin Ligase TRIM40 Attenuates Antiviral Immune Responses by Targeting MDA5 and RIG-I. Cell Rep. 2017, 21, 1613–1623.
- Noguchi, K.; Okumura, F.; Takahashi, N.; Kataoka, A.; Kamiyama, T.; Todo, S.; Hatakeyama, S. TRIM40 promotes neddylation of IKKγ and is downregulated in gastrointestinal cancers. Carcinogenesis 2011, 32, 995–1004.
- Liu, Z.-S.; Zhang, Z.-Y.; Cai, H.; Zhao, M.; Mao, J.; Dai, J.; Xia, T.; Zhang, X.-M.; Li, T. RINCK (TRIM41)-mediated monoubiquitination of cGAS promotes antiviral innate immune responses. Cell Biosci. 2018, 8, 1–9.
- Yang, B.; Wang, J.; Wang, Y.; Zhou, H.; Wu, X.; Tian, Z.; Sun, B. Novel Function of Trim44 Promotes an Antiviral Response by Stabilizing VISA. J. Immunol. 2013, 190, 3613–3619.
- Shibata, M.; Sato, T.; Nukiwa, R.; Ariga, T.; Hatakeyama, S. TRIM45 negatively regulates NF-κB-mediated transcription and suppresses cell proliferation. Biochem. Biophys. Res. Commun. 2012, 423, 104–109.
- Liu, B.; Li, N.L.; Shen, Y.; Bao, X.; Fabrizio, T.; Elbahesh, H.; Webby, R.J.; Li, K. The C-Terminal Tail of TRIM56 Dictates Antiviral Restriction of Influenza A and B Viruses by Impeding Viral RNA Synthesis. J. Virol. 2016, 90, 4369–4382.
- Seo, G.J.; Kim, C.; Shin, W.-J.; Sklan, E.H.; Eoh, H.; Jung, J.U. TRIM56-mediated monoubiquitination of cGAS for cytosolic DNA sensing. Nat. Commun. 2018, 9, 613.
- Tsuchida, T.; Zou, J.; Saitoh, T.; Kumar, H.; Abe, T.; Matsuura, Y.; Kawai, T.; Akira, S. The Ubiquitin Ligase TRIM56 Regulates Innate Immune Responses to Intracellular Double-Stranded DNA. Immunity 2010, 33, 765–776.
- Shen, Y.; Li, N.L.; Wang, J.; Liu, B.; Lester, S.; Li, K. TRIM56 is an essential component of the TLR3 antiviral signaling pathway. J. Biol. Chem. 2012, 287, 36404–36413.
- Kondo, T.; Watanabe, M.; Hatakeyama, S. TRIM59 interacts with ECSIT and negatively regulates NF-κB and IRF-3/7-mediated signal pathways. Biochem. Biophys. Res. Commun. 2012, 422, 501–507.
- Wynne, C.; Lazzari, E.; Smith, S.; McCarthy, E.M.; Joan, N.G.; Kallal, L.E.; Higgs, R.; Greco, A.; Cryan, S.A.; Biron, C.A.; et al. TRIM68 negatively regulates IFN-β production by degrading TRK fused gene, a novel driver of IFN-β downstream of anti-viral detection systems. PLoS ONE 2014, 9, e101503.
- Yu, C.-F.; Peng, W.-M.; Schlee, M.; Barchet, W.; Eis-Hübinger, A.M.; Kolanus, W.; Geyer, M.; Schmitt, S.; Steinhagen, F.; Oldenburg, J.; et al. SOCS1 and SOCS3 Target IRF7 Degradation To Suppress TLR7-Mediated Type I IFN Production of Human Plasmacytoid Dendritic Cells. J. Immunol. 2018, 200, 4024–4035.
- Nakagawa, K.; Yokosawa, H. PIAS3 induces SUMO-1 modification and transcriptional repression of IRF1. FEBS Lett. 2002, 530, 204–208.
- Ng, S.L.; Friedman, B.A.; Schmid, S.; Gertz, J.; Myers, R.M.; tenOever, B.R.; Maniatis, T. IKKe regulates the balance between type I and type II interferon responses. Proc. Natl. Acad. Sci. USA 2011, 108, 21170–21175.
- Everett, R.D.; Boutell, C.; Hale, B.G. Interplay between viruses and host sumoylation pathways. Nat. Rev. Microbiol. 2013, 11, 400–411.
- Hannoun, Z.; Maarifi, G.; Chelbi-Alix, M.K. The implication of SUMO in intrinsic and innate immunity. Cytokine Growth Factor Rev. 2016, 29, 3–16.
- Harty, R.N.; Pitha, P.M.; Okumura, A. Antiviral activity of innate immune protein ISG15. J. Innate Immun. 2009, 1, 397–404.
- Freitas, B.T.; Scholte, F.E.M.; Bergeron, É.; Pegan, S.D. How ISG15 combats viral infection. Virus Res. 2020, 286, 198036.
- El-Asmi, F.; McManus, F.P.; Brantis-de-Carvalho, C.E.; Valle-Casuso, J.C.; Thibault, P.; Chelbi-Alix, M.K. Cross-talk between SUMOylation and ISGylation in response to interferon. Cytokine 2020, 129, 155025.
- Takaoka, A.; Hayakawa, S.; Yanai, H.; Stoiber, D.; Negishi, H.; Kikuchi, H.; Sasaki, S.; Imai, K.; Shibue, T.; Honda, K.; et al. Integration of interferon-à/á signalling to p53 responses in tumour suppression and antiviral defence. Nature 2003, 424, 516–523.
- Jain, A.K.; Allton, K.; Duncan, A.D.; Barton, M.C. TRIM24 is a p53-induced E3-ubiquitin ligase that undergoes ATM-mediated phosphorylation and autodegradation during DNA damage. Mol. Cell. Biol. 2014, 34, 2695–2709.
- Liu, J.; Zhang, C.; Wang, X.; Hu, W.; Feng, Z. Tumor suppressor p53 cross-talks with TRIM family proteins. Genes Dis. 2020.