The Eph receptors represent the largest group among Receptor Tyrosine kinase (RTK) families. The Eph/ephrin signaling axis plays center stage during development, and the deep perturbation of signaling consequent to its dysregulation in cancer reveals the multiplicity and complexity underlying its function. In the last decades, they have emerged as key players in solid tumors, including colorectal cancer (CRC). EphA2 is involved in tumor progression and resistance to therapy.
- EphA2
- EGFR
- TKI
- ephrins
- CRC
- CSCs
- drug resistance
- cetuximab
- intra-tumor heterogeneity
- inter-tumor heterogeneity
1. Introduction
1.1. General Structure of Eph Receptors and Ephrin Ligands
The EphA2 receptor belongs to the Eph (erythropoietin-producing human hepatocellular) superfamily, the largest among tyrosine kinase receptor families [1]. Eph receptors are classified into Eph-A and Eph-B subfamilies depending on their sequence homologies and binding affinity for their cognate ephrin ligands. Although Eph receptors preferentially bind ligands of the same class, cross-binding has been described [for a review, [2,3,4] (Figure 1). All Eph receptors contain an extracellular region, with a conserved N-terminal globular ligand-binding domain (LBD), a cysteine-rich domain which comprises a Sushi and an epidermal growth factor (EGF)-like domain and two fibronectin type-III repeats (FN1 and FN2). The intracellular region contains a juxtamembrane region (JM), a tyrosine kinase domain, a sterile alpha motif (SAM) domain, and a PDZ (Post-synaptic density protein-95, Drosophila disc large tumor suppressor (Dlg), Zona occludens-1) domain-binding motif that are responsible for the interaction with effector molecules. The receptor homo- and hetero-oligomerization involves the extracellular domains (LBD and cys-rich domain) [5,6]. The SAM domain is involved in receptor-receptor interactions, possibly aiding homo- or hetero-oligomerization. The ectodomain and the intracellular domain are linked by a transmembrane helix (TM) [for reviews, [2,3] (Figure 1). Ephrins ligands are divided into ephrin-A and ephrin-B subclasses [5,7]. Ephrin-A proteins (A1–A6) are anchored to the extracellular cell membrane via a glycosyl phosphatidylinositol (GPI) linkage that could be released to activate EphA receptors at distance [8]. Ephrin-B members (B1–B3) are transmembrane proteins containing a cytoplasmic domain with several conserved tyrosine residues and a terminal PDZ-binding motif allowing the interaction with proteins involved in cytoskeleton organization and cell adhesion (Figure 1). Thus, Eph-ephrin signaling is transduced either directly (in the case of ephrin-Bs) or by interaction with intracellular proteins (like Fyn) or other transmembrane proteins (like the neurotrophin receptor p75) (as for ephrin-As [9] (Figure 1).
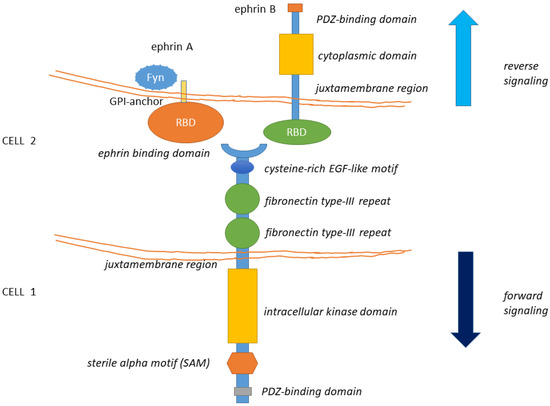
Figure 1. The structure of Eph receptors and their ligands is shown. Eph receptors are consisting of an extracellular structure consisting of an ephrin binding domain connected to two fibronectin type-III repeats by a cysteine-rich EGF-like motif. The juxtamembrane region connects the extracellular portion of the receptor to the intracellular kinase domain that is linked to a sterile alpha motif (SAM) domain and PDZ-binding motif. Eph ligands (ephrin-A/B) are composed of a GPI-anchored receptor binding domain in the case of the ephrin-A type and a receptor-binding domain connected by a juxtamembrane domain to a cytoplasmic domain and a PDZ interaction motif, in the case of ephrin-B. Eph-Ephrin signaling is transduced either directly (in the case of ephrin-Bs) or by interaction with Fyn (as has been observed with ephrin-As). Ligand binding likely initiates clustering, aided by receptor-receptor interactions mediated by the SAM domain and by the PDZ (Post-synaptic density protein-95, Drosophila disc large tumor suppressor (Dlg), Zona occludens-1)-domain-binding motif. The formed complexes mediate bi-directional signaling called ephrin “reverse” and Eph “forward” signaling.
1.2. General Features of Eph-Ephrin Signaling
Since both Eph receptors and ephrins are anchored to the plasma membrane, the Eph-ephrin signaling is intrinsically bidirectional. The forward signaling is consequent to ligand binding and clustering of the receptors on the expressing cells. Trans-phosphorylation of clustered Eph receptors in the juxtamembrane domain enables efficient kinase activity [10,11]. Phosphorylation of the conserved tyrosine in the activation loop appears to be less critical for Eph receptor activation than it generally is for RTKs, mainly contributing to its maximal activity [10,12]. The reverse signaling takes place in the ligand expressing cells [13]. Both repulsive and attractive effects can be consequent to Eph-ephrin binding between cells: additionally, an initial cell-cell adhesion event mediated by the Eph-ephrin interaction may switch to a repulsive one in a time-dependent way, as the effect of cleavage of the membrane-bound ephrin or internalization of the receptor-ligand complex [14]. Ephrins can also attenuate forward signaling by Eph receptors co-expressed in the same cell [15] and also receptor delivery in extracellular vesicles to ligand expressing cancer cells has been shown to be functionally relevant, in cancer settings and in response to stress [16]. The Eph receptors also display non-catalytic functions. For instance, there are two pseudo-kinases (i.e., EphA10 and EphB6) within this large family and their function may be involved in tumorigenesis and resistance to therapy. Such a complexity supports a high adaptive potential, allowing for switching the Eph-ephrin signaling according to changes of both intracellular and extracellular stimuli.
2. EphA2 Signaling
2.1. EphA2 Signaling in Normal Cells
The EphA2 receptor is a 130-kDa transmembrane glycoprotein identified in the early 90′ in Hela cells during a screen for RTKs [18]. EphA2 potentially interacts with any ephrin-A ligand, with the most frequent partner being the membrane-bound, GPI-anchored ephrin-A1, this latter discovered in 1994 [19]. After ligand binding and trans-tyrosine phosphorylation, EphA2 forms a complex with c-Cbl, to be targeted to endosomes and degraded. About 35% of the receptor is recycled back to the plasma membrane [20]. The EphA2 forward signaling is executed through ligand-instigated binding of downstream adaptors and signaling partners [21]. In fact, as for other RTKs, phosphorylation of the tyrosine residues creates docking sites for SH2/SH3 containing-proteins such as Fyn, Src, Nck, Crk, RasGAP, LMW-PTP, PI3K, and the adapter proteins Grb2, Grb10, and SLAP. EphA2 modulates cytoskeletal organization through Rho/Rac GTPases [22]. Most of these proteins affect depolymerization of the actin cytoskeleton while others modulate cell adhesion [23] with important consequences on vascular assembly, angiogenesis, and cell migration [24]. The reverse signaling elicited by EphA2 (acting as a ligand) on the ephrin expressing cell is poorly characterized and known to be mediated, for ephrinAs, by the src family kinase Fyn. Reverse signaling can mediate cell adhesion or repulsion and modulates axon guidance and synaptogenesis in the developing brain [25]. Forward signaling by EphA2 has inhibitory effects on cell proliferation, through Ras/MAPK [26]. Erk inhibition takes place through activation of GAPs and/or inhibition of GEFs [2,27]. EphA2 kinase-dependent signaling thus suppresses the AKT–mTORC1 and RAS–ERK oncogenic pathways and inhibits cell adhesion and migration [28,29] (Figure 2A). For instance, ligand-bound EphA2 attenuated Erk activation in primary keratinocytes and hepatoma cells [30]; Ephrin-A/EphA signaling suppressed Erk activation induced by IGF-1 in myoblasts, facilitating myogenic differentiation [31]. In neurons, EphA-dependent Erk inhibition suppressed the effects of the TrkB RTK on growth cone motility [32,33].
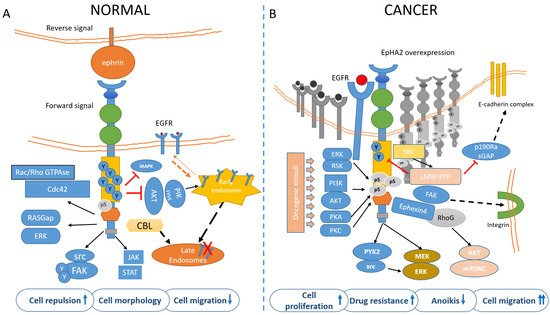
Figure 2. EphA2 signaling in normal (A) and cancer (B) cells. (A) In untransformed cells, EphA2 is engaged by its ligands, mainly EphrinA1 and highly tyrosine-phosphorylated. This mediates cell adhesion/repulsion through activation of Rac/Rho GTPAses and RASGap. Ligand binding also mediates inhibition of MAPK and AKT. Upon ligand binding, the EphA2 is targeted to endosomes in a CBL-mediated process. (B) In cancer cells, unliganded and overexpressed EphA2 is mainly phosphorylated in ser897 by PI3k/AKT, ERK/RSK, PKA, and PKC, in response to oncogenic stimuli The Akt-mTORC1, Raf-MEK-ERK, and Pyk2-Src-ERK signaling pathways were identified as the downstream signaling of the EphA2 non-canonical pathway. S897-phosphorylated EphA2 recruits Ephexin4 that in turn acts on RhoG to promote cell migration and anoikis resistance (this latter effect through a RhoG-AKT pathway). Further, FAK-integrin mediates cell adhesion and migration and may promote CSC features, including drug resistance (please also see Figure 3). The phospho-tyrosine content of EphA2 is also reduced by the LMW-PTPase, frequently overexpressed in cancer. The pro-tumorigenic contribution of EphA2 may thus derive from ligand independency, overexpression, reduced phospho-tyrosine content, and increased serine/threonine phosphorylation. Additionally, ligand-stimulated EphA2 negatively modulates the recycling of EGFR, by inhibiting AKT/PIKfyve, thus reducing the amount of available EGFR on the plasma membrane and migration. On the other hand, such feedback is attenuated in transformed cells, where EGFR levels in the plasma membrane are increased and this correlates with ligand independency of EphA2 and activation of motile responses to EGF.
2.2. EphA2 in Tissue Patterning
We believe that a short journey into the role of EphA2 in tissue patterning and homeostasis may turn useful to better illustrate its involvement in tumor progression, apparently tumor-context specific but obeying signaling principles common to tissue patterning and repair. Eph receptors and ephrins have a key role in cell positioning, cell motility, cell differentiation, control of tissue morphogenesis and patterning, development of the vascular system (for a review, [2,34]). In fact, during tissue patterning, Eph receptors engagement by their ligands impedes cell mixing during tissue development and is essential to create functional topographic domains driving the formation of distinct cellular compartments [35,36,37]. For instance, forward signaling by EphA2 and its ligands aids in establishing synaptic connections in the developing nervous system by modulating growth cone guidance and axon branching [3]. Additionally, Ephrin-A2 reverse signaling inhibited the proliferation of neural progenitor cells, thus negatively modulating neurogenesis [38,39]. In developing mammary glands, EphA2 is important for promoting branching morphogenesis in vivo, as being expressed in mammary progenitor cells [40]. Ephs or ephrins may also cooperate with cell junctional modules (tight junctions and adherens junctions) to facilitate cell sorting processes and preserve the epithelial integrity and physiology in embryonal and adult tissues [41,42]. In normal colon epithelia, several studies have shown a decreasing gradient of EphB2 expression from the base to the top of the crypt, whereas EphA2 expression was observed in the differentiated compartment of the crypt apical columnar cells [43]. In fact, EphA2 is implicated in the repair of the gut epithelia [44] and of kidney epithelia during ischemia-reperfusion injury [45]. All these repair processes imply activation or reactivation of embryonal programs, like EMT or MET. Not coincidentally, as above mentioned, those programs are frequently reactivated in cancer cells [46,47].
3. Molecular Determinants of EphA2 Signaling in Tumors
In all the tumor settings studied, the role of EphA2, which ranges from a tumor-suppressive to a pro-tumorigenic one, depends on a number of intrinsic and extrinsic factors, some of which have been recently determined: its subcellular localization, the levels of expression, the presence of the ligand and the crosstalk with other receptors, such as the Epidermal Growth Factor Receptor (EGFR).
3.1. Intracellular Localization of EphA2
In non-neoplastic epithelia, EphA2 is localized to sites of cell-cell contact, in an E-cadherin-dependent way [48]. In absence of E-cadherin, associated with reduced cell-cell contacts and pro-metastatic behavior of the cancer cells, EphA2 was redistributed to membrane ruffles where it cannot engage with membrane-bound ligand ephrin-A1 on adjacent cells, thus reducing the tumor-suppressive juxtacrine signaling [41,48]. Also in cells lacking Claudin 4, another event associated with acquired pro-tumorigenic potential, EphA2 was increased and mislocalized and this correlated with increased oncogenic signaling [49].
3.2. Expression Levels of EphA2
EphA2 is highly expressed in many cancers with important prognostic implications. Elevated EphA2 expression positively correlated with poor prognosis, improved metastatic potential, and reduced overall survival of patients, in a tumor context-specific functioning (Table 1). Notably, the EphA2 is rarely mutated or amplified in cancer tissues [50,51]. However, expression of EphA2 may be modulated by p53, Ras and negatively modulated by estrogens [48,52,53]. In established cell line cultures, EphA2 expression was higher in cancer cells than in untransformed ones: increased staining intensity was observed, for example, in a large fraction of breast carcinoma cells (an average of 87%) when compared to benign mammary epithelial cells (an average of 3%) [54]. Related to this, overexpression of EphA2 was sufficient to transform immortalized mammary epithelial cells [55]. Additionally, EphA2 is present in GBM cells in a mainly non–tyrosine-phosphorylated state [56].
Table 1. Examples of EphA2 overexpression in human malignancies, with its significance and the number of cases analyzed.
|
Cancer Type |
mRNA Protein |
Linked to |
Cases (n) |
Ref |
|---|---|---|---|---|
|
Esophageal Squamous Cell Carcinoma |
Protein |
loco regional metastases; pathological grade; reduced OS |
80 |
Miyazaki et al., 2002 [57] |
|
Gastric Cancer |
Protein |
cancer recurrence (in association with YAP) |
47 |
Huang et al., 2020 [58] |
|
Prostate cancer |
Protein |
pathological grading |
93 |
Zeng et al., 2003 [59] |
|
Colorectal cancer |
mRNA protein |
CSC markers (CD44 and Lgr5); reduced OS |
338 |
Dunne et al., 2016 [60] |
|
Colorectal cancer |
mRNA |
poor prognosis and response to cetuximab |
226 |
Strimpakos et al., 2013 [61] |
|
Colorectal cancer |
mRNA |
tumor progression and poor OS (EphA2 with miR-423-5p, CREB1, ADAMTS14) |
1663 (TGCA) |
De Robertis et al., 2018 [43] |
|
Colorectal cancer |
mRNA |
worse PFS despite EGFRhigh (cetuximab-treated patients) |
80 (TGCA) |
De Robertis et al., 2017 [62] |
|
Ovarian carcinoma |
Protein |
aggressive features and median survival |
79 |
Thaker et al., 2004 [63] |
|
Ovarian cancer |
mRNA protein |
poor survival |
118 |
Han et al., 2005 [64] |
|
Epithelial Ovarian Cancer |
poor survival (stronger when combined with p53null status) |
79 |
Merritt et al., 2006 [65] |
|
|
Endometrial cancer |
Protein |
higher pathological grade and clinical stage; shorter disease-specific survival (DSS) |
139 |
Merritt et al., 2011 [66] |
|
Cervical carcinoma |
mRNA |
decreased overall survival (OS) |
206 |
Wu et al., 2004 [67] |
|
Head and neck squamous cell carcinoma |
mRNA protein |
higher clinical stage, recurrence, and lymph node metastasis; reduced disease-free survival (DFS) and OS |
98 |
Liu et al., 2011 [68] |
|
Glioblastoma |
mRNA protein |
increased pathological grade; reduced OS |
21 |
Liu et al., 2006 [69] |
|
Malignant glioma |
protein |
decreased DFS and OS (oppositely to EphrinA1) |
78 |
Li et al., 2010 [70] |
|
Glioblastoma multiforme |
protein |
Reduced OS |
40 |
Wang et al., 2008 [71] |
|
Renal Cell Carcinoma |
protein |
increased pathological grade, reduced DFS and OS |
34 |
Herrem et al., 2005 [72] |
|
Renal Cell Carcinoma |
protein |
reduced OS |
62 |
Xu et al., 2014 [73] |
|
Non-Small-Cell-Lung-Cancer |
protein |
smoking history; reduced PFS and OS |
279 |
Brannan et al., 2009 [74] |
|
Non-Small-Cell-Lung-Cancer |
protein |
reduced overall survival (Stronger when associated with PKR) |
218 |
Guo et al., 2013 [75] |
|
Non-Small-Cell-Lung-Cancer |
protein |
brain metastases; reduced OS |
270 |
Kinch et al., 2003 [76] |
|
Hepatocellular carcinoma |
mRNA protein |
higher pathological grade; and reduced OS |
40 |
Cui et al., 2010 [77] |
|
Hepatocellular carcinoma |
protein |
decreased OS |
129 |
Yang et al., 2009 [78] |
|
Gastric cancer |
protein |
higher in high-risk macroscopic grade 3 and 4 tumors |
49 |
Nakamura et al., 2005 [79] |
3.3. Ligand-Dependent EphA2 Signaling
A conspicuous amount of evidence suggests that ligand-mediated activation of EphA2 has tumor-suppressive functions. For instance, inverse expression of ephrin-A1 and EphA2 in human breast cancer cell lines was a frequent finding [80,81]. When tumors were grown in vivo, EphA2 appeared to be poorly activated by the endogenous ephrin-A [29]. Consistent with the previous observations, regulation of EphA2 expression in GBM by Fc-ephrin-A1 stimulation resulted in the loss of self-renewal ability and decreased proliferation in vitro and in vivo [82,83]. Ephrin-A1 ligand-induced EphA2 phosphorylation induces receptor endocytosis and the CBL ubiquitin-ligase mediated proteasome degradation [20,84]. Induction of ephrins may represent per se a mechanism for silencing Eph signaling. For example, during mouse ESC differentiation, FGF4 reduces EphA2 signaling, by transcriptionally inducing its ligands. This correlated with increased tyrosine phosphorylation and reduced Ser/Thr phosphorylation of EphA2 and reduced expression of pluripotency core factors, thereby leading to ESC differentiation [83]. Differently to other RTKs, activation of Eph receptors by ephrins does not increase cell proliferation or transform murine fibroblasts: conversely, it rather inhibited the Ras/MAPK and attenuated mitogen-activated protein kinase (MAPK) activation by platelet-derived growth factor (PDGF), epidermal growth factor (EGF) and vascular endothelial growth factor (VEGF), in a range of cell lines [85]. In cancer cells, including PTEN deficient prostate cancer cells and glioma cells, ephrin-dependent EphA2 activation led to rapid dephosphorylation of Akt at T308 and S473 residues leading in some cases to mTORC1 inactivation and decreased cell growth and migration [86,87,88] (Figure 2A). However, the effect of ephrin-A1 on EphA2 expressing cells may also be cell type-specific and transformation status-dependent: for instance, ephrin-A1 treatment inhibited proliferation of prostate cancer cells but failed to do so in fibroblasts [85]. Progranulin, a recently discovered EphA2 ligand, induced transient activation of MAPK in both untransformed HUVECs and transformed prostate cancer cells, but sustained activation of AKT was observed only in the latter cancer cells [89]. Altogether, this suggests that the dichotomic view (tumor suppression vs tumor promotion based on ligand availability) is too simple. Ephrin-driven forward signaling suppressed AKT activation in an ephrin-dependent way [86,87,88] ligand-bound EphA2 suppressed the recycling of EGFR to the plasma membrane, causing EGFR accumulation at the endosomes and thereby attenuating EGFR-induced cell migration. This happened in both Mouse Embryo Fibroblasts (MEFs) and in triple-negative breast cancer cells (MDA-MB-231) and was due to reduced PIKfyve activation in early endosomes following EphA2-mediated inhibition of AKT [90,91] (Figure 2A). In keeping with a tumor-suppressive role for ligand-bound EphA2, forward signaling elicited by ephrin-A ligands from normal cells on EphA2 expressing, RasV12 positive cells caused repulsion and segregation of the transformed cells [92].
3.4. Ligand-Independent Activation of EphA2
Low juxtacrine signaling and/or insufficient levels of ephrinA1 on cancer cells reduce EphA2 tyrosine phosphorylation [56] and this leads to attenuated internalization and degradation of EphA2 receptor, with a relative increase of EphA2 levels. Concomitantly, when the ephrinA1-mediated inhibition of AKT is removed, EGFR recycling to the plasma membrane is reduced and the EphA2 ligand-independent effect is switched on by phosphorylation on S897 (Figure 2B). Phosphorylation of the S897 residue (among the 25 ser/thr residues in EphA2) in the region linking the kinase domain with the SAM domain is the main target for “non-canonical” ephrin-independent and/or kinase-independent EphA2 signaling [87] and this activated multiple mechanisms, encompassing the downstream activation of Akt–mTORC1, Raf–MEK–ERK, and Pyk2–Src–ERK [93] (Figure 2B). Additionally, the association between EphA2 and FAK resulted in integrin-mediated adhesion, cell spreading, and migration [94] (Figure 2B). Further, unliganded, EphA2 destabilized adherent junctions via Rho-GTP activation, by inhibiting p190 RhoGAP (a Rho-GTP inhibitor) through activating the low molecular weight phospho-tyrosine phosphatase (LMW-PTP) [95]. LMW-PTP by itself may decrease the phospho-tyrosine content of EphA2 [96], possibly when activated by stress signals [97] (Figure 2B). LMW-PTP, overexpressed in many cancers, has overlapping functions with EphA2, including cell motility and resistance to therapy [98,99]. S897-phosphorylated EphA2 recruited Ephexin4 to promote cell migration and anoikis resistance via RhoG and Rac [100]. RhoG may also activate the PI3K/Akt signaling pathway to promote cell proliferation and survival independently of the activation of Rac [101,102]. RhoG-mediated activation of PI3K and Akt also suppressed anoikis [103]. Anoikis is an apoptotic modality induced by the detachment of adherent cells from the extracellular matrix and its suppression is a feature of metastatic cells [104] (Figure 2). Phosphorylation of the S897 residue in the region linking the kinase domain with the SAM domain is thus the main target for the “non-canonical” ephrin-independent and/or kinase-independent EphA2 signaling [87]. AKT, RSK, PKA, and PKC phosphorylated EphA2-S897, and this increased cell migration/invasion and metastasis and promoted cancer stem cell-like features [105,106,107] (Figure 2B). Structurally, unliganded EphA2 forms predominantly dimers rather than high-order oligomeric structures [108]. There is also evidence that the unliganded EphA2 receptor JM + kinase region may interact with phosphatidylinositol phosphates (PIPs), even if the physiological relevance of this remains to be addressed [109].
3.5. Tumor Context Modulates EphA2 Signaling
Besides these general mechanisms, EphA2 is endowed with tumor-context specific functions, described below. In gastric cancer cell lines, ligand-independent EphA2 activation upregulated N-cadherin and Snail, and the Wnt/β-catenin targets TCF4, Cyclin-D1, and c-Myc, thereby triggering epithelial-to-mesenchymal transition (EMT) [110]. EMT is a complex process during which tumor cells progressively acquire mesenchymal features (such as resistance to stress and acquisition of migratory ability and metabolic resilience). In detail, overexpressed EphA2 was shown to bind to Wnt-1 and to promote beta-catenin nuclear accumulation. This upregulated c-MYC that, in turn, promoted further EphA2 increase in a feed-forward manner, by binding to the EphA2 promoter [111]. As mentioned before, in breast cancer cells Ephexin4, a guanine nucleotide exchange factor (GEF) for RhoG, interacted with S897-phosphorylated EphA2 and mediated ephrin-independent cell migration, invasion, and resistance to anoikis (Figure 2B). In glioblastoma (GBM), stimulation of the cells with EGF induced MEK- and RSK-dependent EphA2 S897 phosphorylation [112]. Miao and coworkers found that EphA2 S897 phosphorylation was present mainly in grade IV human glioma specimens, in regions enriched for pS473-Akt signal and invasive cells [87]. S897 phosphorylation of EphA2 has also been involved in determining the aggressiveness of thyroid cancer cells and shown to be mediated by ERK1/2 activation downstream of oncogenes like RET (RET/PTC), KRAS (G12R), or BRAFV600E [113]. The same EphA2 residue is phosphorylated by ionizing radiation in a MEK/ERK/RSK-dependent manner, mediated by increased ROS, in multiple cancer cell lines [114].
Regarding the events downstream of EphA2 S897, the Akt–mammalian target of ra-pamycin complex 1(mTORC1), Raf–MEK–ERK, and Pyk2–Src–ERK pathways were shown to be downstream effectors of the S897 EphA2 pathway in cholangiocarcinoma cells [115]. In prostate cancer and GBM, EphA2 S897 expression induced amoeboid motil-ity, which correlated with the induction of stemness markers, increased clonogenic poten-tial and tumour growth [82,116,117]. The EphA2 S897 increased in glucose starvation conditions in GBM cells and this correlated with cell survival and ROS-mediated ERK-RSK activation, induced by the cystine/glutamate antiporter xCT [118]. Thus, the S897 phosphorylation of EphA2 may work as a stress rheostat, transducing adaptive responses and thereby influencing tumor progression.
Notably, the “simple” abrogation of tyrosine phosphorylation in EphA2 may repre-sent “per se” an oncogenic signal. For instance, reintroduction of pY772A EphA2 in EphA2 knock-down naso-pharyngeal-carcinoma (NPC) cells increased cell proliferation, anchorage-independent growth in vitro and tumor growth in vivo. Mechanistically, EphA2-Y772A triggered activation (rather than inhibition) of Shp2/Erk-1/2 signaling pathway in the NPC cells, the latter involving binding of GAB1 and GRB2 as well [119]. In support of this, expression of kinase-deficient variants of EphA2 in breast cancer cells led to decreased tumor volume and increased tumor cell apoptosis [120].
A number of EphA2 mutations interfering with ephrin binding or kinase activity in cancer tissues such as intrahepatic cholangiocarcinoma (ICC) is being growingly recognized [121]. For instance, an EphA2 A859D Y772 dead mutant, exhibiting lower levels of phosphorylated Y772 and suppressed degradation through CBL, was recently identified in squamous cell carcinoma (SSC) and malignant pleural mesothelioma (MPM) speci-mens [122]. Contrariwise, tyrosine kinase activity of overexpressed EphA2 was also shown as required for the S897 phosphorylation via ERK to stimulate GBM cell prolifera-tion [112], thus showing the limit of a dichotomist view of canonical vs non-canonical EphA2 signaling and suggesting a more complex scenarios where both signaling modalities are highly interconnected [see also [28]]. For example, in Ewing sarcoma (ES) EphA2 promoted angiogenesis via ligand- (and caveolin-1)-dependent signaling [123], while enhancing tumorigenicity, migration and invasion in vitro and in vivo, in an S897 dependent manner [124].
EphA2 promotes resistance to a broad range of anticancer agents, including conventional agents like cisplatin and paclitaxel, EGFR Tyrosine Kinase inhibitors, BRAF inhibitors and EGFR-blocking antibodies. It does so by activating tumor speciifc pathways and by impinging on key processes such as the Epithelial to Mesenchymal Transition (EMT) (see cover figure, please).
This entry is adapted from the peer-reviewed paper 10.3390/cancers13040700