Chronic kidney disease (CKD) is one of the major health problems of the modern age. It represents an important public health challenge with an ever-lasting rising prevalence, which reached almost 700 million by the year 2017. Therefore, it is very important to identify patients at risk for CKD development and discover risk factors that cause the progression of the disease. Several studies have tackled this conundrum in recent years, novel markers have been identified, and new insights into the pathogenesis of CKD have been gained. This review summarizes the evidence on markers of inflammation and their role in the development and progression of CKD. It will focus primarily on cytokines, chemokines, and cell adhesion molecules. Nevertheless, further large, multicenter studies are needed to establish the role of these markers and confirm possible treatment options in everyday clinical practice.
- inflammation markers
- chronic kidney disease
- cytokines
- chemokines
- cell adhesion molecules
1. Introduction
Chronic kidney disease (CKD) represents a condition with reduced renal function, which is defined by glomerular filtration rate (GFR) < 60 mL/min per 1.73 m2 or with present markers of kidney damage or both for more than 3 months and with implications for health. Markers of kidney damage are defined by Kidney Disease Improving Global Outcomes (KDIGO) guidelines from 2012 as albuminuria, urine sediment abnormalities, electrolyte, and other abnormalities due to tubular disorders, abnormalities detected by histology, structural abnormalities detected by imaging, and history of kidney transplantation. CKD should be categorized into categories according to GFR ranging from G1–G5 (GFR in G1 ≥ 90, G2 60–89, G3a 45–59, G3b 30–44, G4 15–29, and G5 ≤ 15 mL/min/1.73 m2), and albuminuria should be accounted for in categories from A1–A3 (albumin excretion rate in A1 < 30, A2 30–300, and A3 > 300 mg/24 h) [1]. The pathophysiology of CKD is extremely complex with its clinical course dependent on a broad spectrum of different etiologies all leading towards kidney failure. The main causes of CKD in all high-income and middle-income countries are diabetes mellitus (DM) and arterial hypertension (AH) [2]. The global prevalence of all-stage CKD recorded in 2017 was 697.5 million or 9.1%, and 1.2 million people died from CKD in 2017 [3]. Patients are often asymptomatic or have non-specific symptoms such as loss of appetite, pruritus, and fatigue, so diagnosis is usually achieved by estimation of GFR using several available equations, or less frequently by measurement of GFR via exogenous markers. Additionally, a diagnosis can be made by performing a kidney biopsy, which can commonly show glomerular sclerosis, tubular atrophy, and interstitial fibrosis [2]. One of the hallmarks of CKD resulting in its development, progression, and complications is persistent, low-grade inflammation [4][5].
Inflammation is a natural and necessary body response to different stimuli. It is responsible for the migration of immune system cells to the stimuli target site following a series of steps facilitated and coordinated by cytokines, chemokines, and acute-phase proteins. In the acute setting, this provides a resolution of the problem and enables the return to the status quo. Chronic inflammation on the other hand can lead to tissue damage and fibrosis. As such it has been associated with numerous diseases, CKD included [6]. We are only beginning to understand this very fine-tuned mechanism and research is still ongoing. Until now, several different pathways of inflammation response have been discovered, such as p38 mitogen-activated protein kinase (p38 MAPK), interleukin-6 (IL-6)/Janus kinase (JAK)/signal transducer and activator of transcription 3 (STAT3), and phosphoinositide 3-kinase (PI3K) [7]. (Figure 1).
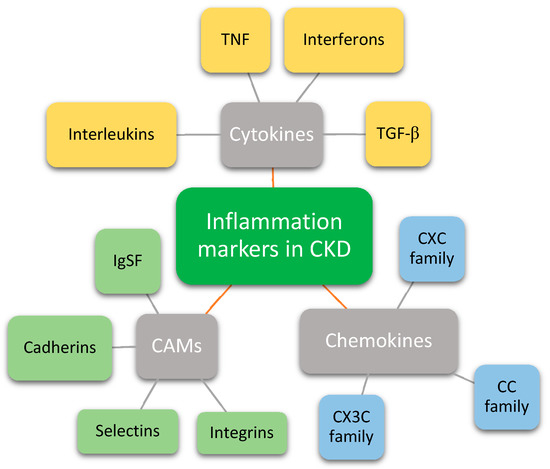
Figure 1. Groups of inflammation markers that have a role in chronic kidney disease. CKD—Chronic kidney disease; TNF—Tumor necrosis factor; TGF-β—Transforming growth factor beta; CAMs—Cell adhesion molecules; IgSF—Immunoglobulin superfamily.
2. Cytokines
Cytokines are small proteins (15–20 kDa) involved in the development and activity of the immune system. They have an important effector and messenger role with involvement in autocrine, paracrine, and endocrine signaling [8][9]. Their short half-life suggests they are usually rapidly eliminated to ensure limited bioactivity but can exhibit systemic effects if necessary. The acute phase is usually short term and sufficient to resolve the injury. Persistent inflammation, either due to prolonged stimulation by biological, chemical, or physical stimuli, or inappropriate reaction against self-antigens, however, can prove to be problematic, as it can lead to development of chronic inflammation [6]. Possible association of cytokines with diabetic nephropathy was already observed almost 30 years ago [10].
2.1. Interleukins
Interleukins are a group of cytokines first seen to be expressed by leukocytes. Due to receptor complexes that interact with various interleukins, they have been divided into families, the most important being: IL-1 superfamily, IL-6 family, IL-10 family, IL-12 family, and IL-17 family [8][9][11][12][13].
2.2. Tumor Necrosis Factor
TNF is another cytokine released by immunologic cells, tumor necrosis factor alpha (TNF-α) specifically by monocytes/macrophages. The TNF ligand family is coded by 18 known human genes; its relative the TNF/nerve growth factor (NGF) is coded by an additional 29 genes. However, not only ligands are important in tissue homeostasis, but their tumor necrosis factor receptors (TNFR) as well. Approximately 30 members of TNFR have been identified, which are type I transmembrane proteins that can be classified into three groups: death receptors, TNFR-associated factor, and decoy receptors [14][15]. These ligands and receptor families have several known functions: control of cell death, orchestration of inflammation, tissue modeling, control of adaptive immunity, and several less understood mechanisms tied to control of brain function, such as perception, fever, anorexia, sleep, etc. [15][16]. They also have an important role in autoimmunity, cancer, infectious disease, and graft-versus-host disease [15]. One of the possible ways it can act on CKD is by raising FGF23, which regulates phosphate homeostasis, causes mineral and bone disorder, and is associated with all-cause mortality of CKD patients [17].
Studies show that TNF-α levels are higher in diabetic patients compared to healthy people, with its levels rising with worsening diabetic nephropathy [18]. Higher soluble TNFR’s were also associated with diabetic kidney disease, where it independently predicted incident cardiovascular disease and mortality [19]. Additionally, their circulating TNFR levels indicated eGFR decline, as well as progression to ESRD [20]. A study on mice has revealed that TNF-α blockade in diabetic nephropathy confers kidney protection visible by reduction of albuminuria, serum creatinine, histopathologic changes, and macrophage recruitment to kidneys [21].
As TNF’s role in diabetic nephropathy has been established, other renal diseases were investigated as well. Oh et al. have reported that higher levels of circulating TNFR are associated with renal function decline as well as worse histopathologic findings in patients with IgAN. Additionally, baseline values predicted subsequent renal progression [20]. TNFR’s effect on declining kidney function was also reported in the Heart and Soul study [22]. Similar findings were reported for TNF-α, where higher serum levels proved to be a potential biomarker for evaluating disease severity as they were linked with proteinuria and renal function decline [23]. Interestingly, however, TNF-α inhibitors have been identified as a potential cause for IgAN, which was reported for use of adalimumab, but not for infliximab [24]. Nonetheless, their use in patients with rheumatoid arthritis from the KOBIO registry did not influence any change in renal function [25]. Furthermore, a study on rats has shown that a combination of empagliflozin, a sodium-glucose co-transporter 2 (SGLT2) inhibitor, and infliximab prevented renal fibrosis to a greater extent than monotherapy, which could present a promising therapeutic option [26].
Genetic studies have revealed that gene polymorphisms of TNF-α are risk factors for the development of nephrotic syndrome among Egyptian children and are linked with steroid resistance [27]. Additionally, some TNF gene polymorphisms in Mexican patients were associated with systemic lupus erythematosus (SLE) and lupus nephritis [28].
2.3. Interferons
IFNs are a class of cytokines first identified due to their property to interfere with viral replication in the host. They have a versatile function ranging from the regulation of immune response, which is associated with autoimmunity, to oncogenesis. They are divided into three types. Type I, such as interferon alpha (INF-α) and interferon beta (IFN-β), are expressed by innate immune cells; type II, also termed interferon gamma (IFN-γ) is expressed by natural killer cells and T lymphocytes; and type III, also termed interferon lambda (IFN-λ) or initially IL-28 and IL-29, are distributed in tissue and act on epithelial surfaces. Type I IFNs bind to the IFN alpha and beta receptor subunit 1 and 2 (IFNAR1 and IFNAR 2), which signal through recruitment of tyrosine kinase 2 (TYK2) and JAK1, respectively. The same signal cascade is activated in IFN type III signaling, however, IFN- λ binds to IL28-R and IL-10R2. IFN-γ binds to the IFN-γ receptor 1 and 2 (IFNGR1 and IFNGR2), which signals through JAK1 and JAK2, respectively [29].
IFN’s role in CKD is less well understood. Undoubtedly it plays an important part in several extrarenal viral infections such as hepatitis C infection, which can also trigger membranoproliferative glomerulonephritis, however, it was also observed in IgAN, lupus nephritis, and renal vasculitis [30]. Grzegorzewska et al. reported that plasma IFN-λ levels in hemodialysis patients are associated with a better response to hepatitis B virus (HBV) vaccine and are independently associated with survival [31].
Several case reports have been described, where interferon was used as a treatment option, mostly for non-renal disease. A patient with immunoglobulin M (IgM) nephropathy presented with nephrotic syndrome and achieved long-term remission with IFN-α [32]. However, plasma levels of IFN-α were reported higher in patients with IgAN by Zheng et al., where they were associated with proteinuria and histologic changes [33]. Similarly, genetic studies reported an association of serum levels of IFN-γ and IgAN and suggested that IFN-γ polymorphisms may be involved in the development and progression of IgAN in the Chinese population [34]. Furthermore, a case report of a patient with hypereosinophilia syndrome who was treated with recombinant IFN-α-2b described he developed progressive renal failure and nephrotic proteinuria 1 year after IFN treatment. Cytokine therapy discontinuation led to a reversal of kidney injury [35]. Similar findings were observed in a patient with multiple sclerosis (MS), where therapy with IFN-β led to the development of nephropathy with nephrotic syndrome [36]. These findings can somewhat be explained with a study by Migliorini et al., who reported that INF-α and IFN-β both have a synergistic, yet different, effect on podocytes and parietal epithelial cells that causes glomerulosclerosis [37]. Due to known systemic adverse effects of IFN administration, some researchers have tried a different approach, using specific drug targeting on mice models. They targeted platelet-derived growth factor receptor β (PDGFRβ)-expressing myofibroblasts using IFN-γ, which attenuated renal fibrosis in obstructive nephropathy [38].
2.4. Transforming Growth Factor Beta
TGF-β is a cytokine involved in the regulation of cell proliferation. Regardless of its name, it can either stimulate or inhibit cell growth and is critical for maintaining immune cell homeostasis. Additionally, it is implicated in the pathogenesis of several diseases such as connective tissue disorders, fibrosis, and cancer. During research of the genome sequence, 33 proteins have been recognized, which now comprise the TGF-β family. They have different receptors and intracellular effectors named Smad proteins, which mediate intracellular signaling and result in regulation of expression of target genes. Gene mutations associated with TGF-β regulation and release are implicated in connective tissue disorders and skeletal disease [39][40][41].
Some CKDs such as membranous and diabetic nephropathy frequently result in ESRD due to renal fibrosis. One of the major regulators appears to be TGF-β, which induces epithelial to mesenchymal transition, up-regulates matrix protein synthesis, inhibits matrix degradation, and alters cell-to-cell interaction [42][43]. One of the pathways that is regulated by TGF-β and is involved in tissue fibrosis by scar formation by myofibroblasts is TGF-β/Mothers against decapentaplegic homolog 3 (SMAD3), additionally, non-canonical pathways such as src kinase, epidermal growth factor receptor (EGFR), JAK/STAT3, and p53 collectively drive the fibrosis process [44][45].
A meta-analysis revealed that elevated serum levels of TGF-β are present in diabetic patients, which poses a high risk for nephropathy [46]. Therefore, a study on mice evaluated the use of pyrrole-imidazole (PI) polyamide, promoter inhibitors targeting TGF-β1, which resulted in improvement of podocyte injury [47]. However, a phase 2 study tried to address the effect of TGF-β1 monoclonal antibody (LY2382770) use in patients with diabetic nephropathy on the renin–angiotensin–aldosterone system (RAAS) inhibitor treatment. The treatment has not resulted in slowing renal function decline [48]. Nonetheless, potential therapeutic targets for progressive renal fibrosis involved in the TGF-β/SMAD3 pathway recently discovered are long non-coding ribonucleic acid (lnc-RNA) Erbb4-IR and lnc-TSI [49][50]. TGF- β/SMAD3-driven fibrosis also represents the pathophysiologic basis of obstructive nephropathy. Recently, a small molecule called petchiether A was identified as a potential inhibitor of SMAD3 phosphorylation, which protects against renal inflammation and fibrosis [51]. A further molecular mechanism is the nuclear factor erythroid 2-like 2 (Nrf2)/SMAD7 pathways, which was targeted recently by bardoxolone, an activator of NRF-2, and resulted in amelioration of tubular necrosis and interstitial fibrosis in mice [52].
3. Chemokines
Chemokines (the term is derived from chemotactic cytokines) are a family of small peptides (8 to 12 kDa) that mediate various processes such as chemotaxis, leucocyte degranulation, hematopoiesis, and angiogenesis. They function by interacting with the cell surface G-protein coupled receptors (GPCRs) and are involved in several pathologic disorders such as cancer, infectious disease, and atherosclerosis. One of recent strategies to tackle these diseases has therefore been the direct inhibition of GPCRs but development of effective chemokine antagonists is elusive. Depending on the spacing of disulfide bridges between peptides, they are classified into four subfamilies: the CXC subfamily (α-subfamily), CC subfamily (β-subfamily), C or XC subfamily (γ-subfamily), and CX3C subfamily (δ-subfamily) [53][54].
3.1. CXC Subfamily
As genome-wide association studies (GWASs) revealed an association of locus in the CXCL12 (CXC ligand 12), also known as stromal cell-derived factor 1 alpha and beta (SDF-1α and β), region with myocardial infarction (MI), a study was done to assess this clinically. In CKD patients from the CRIC study, higher plasma levels of CXCL12 were associated with cardiovascular disease risk factors and incident MI and death [55]. Another study by Klimczak–Tomaniak reported an association of higher CXCL12 levels with left ventricular mass and 24 h blood pressure in CKD patients [56]. However, Chen et al. investigated the role of CXCL12/CXCR4 (CXC receptor 4) pathways in renal vascular development and observed that local CXCL12/CXCR4 signaling preserves microvascular integrity and prevents renal fibrosis. Additionally, this effect was observed after treatment with an angiotensin converting enzyme (ACE) inhibitor [57]. Ribeiro et al. reported that CXCL12 and CXCL8, also known as IL-8, could be used as a marker of vascular damage and dysfunction in uremia [58]. Similar findings were reported by Bouabdallah et al., where they observed that CXCL8 could have a role in vascular calcification in uremia by a mechanism of endothelial cells and vascular smooth muscle cells interaction [59]. CXCL8, alongside with CXCL1, also known as growth-related oncogene-α (GRO-α), and CXCL6, also known as granulocyte chemotactic protein (GCP), were significantly higher in patients with nephrotic syndrome compared to controls and their levels decrease with remission, which could suggest that they play a role in the pathogenesis of nephrotic syndrome [60]. Another chemokine named CXCL16, a scavenger receptor for oxidized low-density lipoprotein (LDL), was reported by Lin et al. to be independently associated with a change in renal function in all-stage CKD [61]. Additionally, it appears to be a marker of renal injury in patients with type 2 DM, where it is independently predictive of microalbuminuria [62][63] and its urinary levels could reflect the degree of interstitial fibrosis and tubular atrophy in patients with advanced diabetic kidney disease [64]. It appears to also play a role in the etiopathogenesis of preeclampsia, where its blockade could pave the way to the discovery of new treatments of preeclampsia [65]. CXCL10, also known as interferon-gamma-inducible protein 10 (IP-10), has shown its value in lupus patients. Urinary levels of CXCL10 were correlated with renal activity score, 24-h proteinuria, and SLE disease activity index (SLEDAI) score. This was also proven for serum CXCL10 and CCL2 levels, however, only urinary levels of CXCL10 indicated renal activity [66][67].
3.2. CC Subfamily
Plasma CCL2, also known as monocyte chemoattractant protein 1 (MCP-1), increases with lowering renal function, however, it appears to be an independent risk factor for death caused by other factors than atherosclerosis in patients with CKD [68]. Nonetheless, CCL2 levels were associated with dyslipidemia in hemodialysis patients [69]. CCL2 baseline values have also shown to be significantly associated with incident CKD [70]. Additionally, higher values of CCL2 reflect tubular injury in patients with diabetic nephropathy [71]. Gene polymorphism studies demonstrated that CCL2 polymorphisms (MCP-1 2518 A > G) may be associated with the risk of developing CKD [72]. Serum and urinary CCL2 were evaluated in pediatric CKD patients compared to controls. Higher levels were observed in CKD patients, with a difference in urinary levels between different CKD etiologies; patients with glomerular disease had higher levels than those with anomalies of the kidney and urinary tract [73]. Additionally, fractional excretion of CCL2 as a marker of inflammation was shown to precede tubular dysfunction in pediatric CKD patients [74]. CCL18, also known as the pulmonary and activation-regulated chemokine, was shown to drive renal inflammation in ANCA-associated crescentic glomerulonephritis and could serve as a biomarker for disease activity and relapse [75].
In biopsy specimens of patients with IgAN, CCL2, and CCL5, also known as regulated upon activation normal T-cell expressed and secreted (RANTES), as well as CCR5 were up-regulated and could play a role in renal fibrosis [76]. The possible role of CCL2 in renal fibrosis and worsening renal function was also highlighted following a systematic review [77]. Interestingly, CCL2 has shown kidney protective properties during acute inflammatory response after renal ischemic/reperfusion injury in mice [78]. Additionally, CCL5, known for its role in the recruitment of macrophages and T lymphocytes into injured tissues, has shown a protective role in a mice model of kidney injury by constraining the proinflammatory actions of CCL2 [79]. Nonetheless, a review of CCL2 regulation has shown that CCL2 blockade in several models of renal disease has ameliorated the disease [80].
3.3. CX3C Subfamily
CX3CL1, also known as fractalkine, has been involved in several kidney diseases, however, studies show conflicting results regarding its disease-promoting or protecting effect [81]. Nonetheless, recent mice studies show that it could play an essential role in lupus nephritis with the development of tubulointerstitial lesions through activation of the Wnt/β-catenin signaling pathway [82]. Additionally, in vitro studies further evaluated the CX3CL1 and Wnt/β-catenin signaling pathway to find that it is associated with podocyte damage and could account for progressive AKI [83]. In Chinese patients with IgAN, plasma CX3CL1 levels were associated with renal function, proteinuria, and histologic changes [84]. Furthermore, they suggest a possible role in the pathogenesis and activity of lupus nephritis [85]. However, a study on mice showed that CX3CR1 inhibition in hypertension could promote hypertensive renal injury [86]. A study investigating CX3CR1 V249I A/G gene polymorphism in CKD patients revealed its possible role in AH, DM, and atherosclerosis [87].
4. Cell Adhesion Molecules
CAMs have a central role in cell communication and connection. They are divided into five groups: integrins, selectins, cadherins, members of the immunoglobulin superfamily (IgSF), and others such as mucins. They differ between one another by structure and ligand binding, as well as the type of binding. Selectins are further divided into P- (platelet), E- (endothelial cells), and L- (leukocyte) selectins. Cadherins, derived from calcium-dependent adherent proteins, can be subdivided into classical cadherins (type I and II), protocadherins, and atypical cadherins. The largest family is the IgSF, which also includes the major histocompatibility complex (MHC) class I and II, and the proteins of the T cell receptor (TCR) complex, such as intracellular adhesion molecules (ICAMs) vascular cell adhesion molecules (VCAMs), mucosal addressin cell adhesion molecule (MAdCAM-1), and activated leukocyte cell adhesion molecule (ALCAM) [88]. The most important CAMs associated with atherogenesis identified until now are VCAM-1, ICAM-1, platelet endothelial cell adhesion molecule 1 (PECAM-1), E-selectin, and P-selectin. As they were identified, research was ongoing about designing treatment modalities by targeting specific CAMs to be able to deliver drugs directly to plaque site [89].
In CKD, several endothelial dysfunction biomarkers, such as ICAM, VCAM, and E-selectin are increased [90]. Additionally, the uremic milieu can increase the levels of endothelial microparticles, which are lined with several adhesion molecules, such as cadherins, ICAM, E-selectin, and integrins, which cause a proinflammatory state, and are related to the development of atherosclerosis [91]. Bevc et al. showed that intima-media thickness (IMT) correlates with serum VCAM-1 in hemodialysis patients [92]. A study by Feng et al. showed that decreasing eGFR is associated with higher VCAM as inflammation in CKD stimulates glomerular expression of VCAMs [93]. In adult-onset minimal change disease, levels of VCAM and E-selectin were increased and associated with severity of nephrotic syndrome [94]. A multicenter study revealed that IgAN and focal segmental glomerulosclerosis (FSGS) had higher levels of P-selectin than healthy controls [95]. However, in CKD patients with atrial fibrillation, levels of P-selectin and E-selectin did not correlate with eGFR [96]. In Asians with type 2 DM, VCAM, but not ICAM, was associated with prevalent diabetic kidney disease and renal function decline [97]. However, Karimi et al., showed that ICAM levels were higher in patients with type 2 DM compared to healthy patients and that higher levels were also associated with more microalbuminuria [98]. As one of the mechanisms of development of diabetic nephropathy is endothelial to mesenchymal transition, studies show that E-cadherin levels fall with more transition and loss of endothelium and could be considered a novel biomarker of diabetic nephropathy development and progression [99]. Similar findings with lower expression of E-cadherin were also observed in children with IgAN [100]. Further studies on diabetic nephropathy revealed that podocyte detachment in the early stage of the disease is mediated through α3β1-integrin [101]. Also, the use of integrin antagonists in a rat model of diabetic nephropathy suggested that treatment with MK-0429 might have a meaningful impact on proteinuria and prevent fibrosis in diabetic nephropathy [102].
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines9020182
References
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2013, 3, 5–14.
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252.
- Global, regional, and national burden of chronic kidney disease, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733.
- Mihai, S.; Codrici, E.; Popescu, I.D.; Enciu, A.M.; Albulescu, L.; Necula, L.G.; Mambet, C.; Anton, G.; Tanase, C. Inflammation-Related Mechanisms in Chronic Kidney Disease Prediction, Progression, and Outcome. J. Immunol. Res. 2018, 2018, 2180373.
- Hojs, R.; Ekart, R.; Bevc, S.; Hojs, N. Markers of Inflammation and Oxidative Stress in the Development and Progression of Renal Disease in Diabetic Patients. Nephron 2016, 133, 159–162.
- Germolec, D.R.; Shipkowski, K.A.; Frawley, R.P.; Evans, E. Markers of Inflammation. Methods Mol. Biol. 2018, 1803, 57–79.
- Yeung, Y.T.; Aziz, F.; Guerrero-Castilla, A.; Arguelles, S. Signaling Pathways in Inflammation and Anti-inflammatory Therapies. Curr. Pharm. Des. 2018, 24, 1449–1484.
- Rose-John, S. Interleukin-6 Family Cytokines. Cold Spring Harb. Perspect. Biol. 2018, 10, a028415.
- Yazdi, A.S.; Ghoreschi, K. The Interleukin-1 Family. Adv. Exp. Med. Biol. 2016, 941, 21–29.
- Hasegawa, G.; Nakano, K.; Sawada, M.; Uno, K.; Shibayama, Y.; Ienaga, K.; Kondo, M. Possible role of tumor necrosis factor and interleukin-1 in the development of diabetic nephropathy. Kidney Int. 1991, 40, 1007–1012.
- Wei, H.; Li, B.; Sun, A.; Guo, F. Interleukin-10 Family Cytokines Immunobiology and Structure. Adv. Exp. Med. Biol. 2019, 1172, 79–96.
- Tait Wojno, E.D.; Hunter, C.A.; Stumhofer, J.S. The Immunobiology of the Interleukin-12 Family: Room for Discovery. Immunity 2019, 50, 851–870.
- Monin, L.; Gaffen, S.L. Interleukin 17 Family Cytokines: Signaling Mechanisms, Biological Activities, and Therapeutic Implications. Cold Spring Harb. Perspect. Biol. 2018, 10, a028522.
- Williams, A.; Wang, E.C.; Thurner, L.; Liu, C.J. Review: Novel Insights Into Tumor Necrosis Factor Receptor, Death Receptor 3, and Progranulin Pathways in Arthritis and Bone Remodeling. Arthritis Rheumatol. 2016, 68, 2845–2856.
- Vanamee, É.S.; Faustman, D.L. Structural principles of tumor necrosis factor superfamily signaling. Sci. Signal. 2018, 11, eaao4910.
- Wallach, D. The Tumor Necrosis Factor Family: Family Conventions and Private Idiosyncrasies. Cold Spring Harb. Perspect. Biol. 2018, 10, a028431.
- Egli-Spichtig, D.; Imenez Silva, P.H.; Glaudemans, B.; Gehring, N.; Bettoni, C.; Zhang, M.Y.H.; Pastor-Arroyo, E.M.; Schőnenberger, D.; Rajski, M.; Hoogewijs, D.; et al. Tumor necrosis factor stimulates fibroblast growth factor 23 levels in chronic kidney disease and non-renal inflammation. Kidney Int. 2019, 96, 890–905.
- Li, X.; Wu, T.T.; Chen, J.; Qiu, W. Elevated expression levels of serum insulin-like growth factor-1, tumor necrosis factor-α and vascular endothelial growth factor 165 might exacerbate type 2 diabetic nephropathy. J. Diabetes Investig. 2017, 8, 108–114.
- Carlsson, A.C.; Östgren, C.J.; Nystrom, F.H.; Länne, T.; Jennersjö, P.; Larsson, A.; Ärnlöv, J. Association of soluble tumor necrosis factor receptors 1 and 2 with nephropathy, cardiovascular events, and total mortality in type 2 diabetes. Cardiovasc. Diabetol. 2016, 15, 40.
- Oh, Y.J.; An, J.N.; Kim, C.T.; Yang, S.H.; Lee, H.; Kim, D.K.; Joo, K.W.; Paik, J.H.; Kang, S.W.; Park, J.T.; et al. Circulating Tumor Necrosis Factor α Receptors Predict the Outcomes of Human IgA Nephropathy: A Prospective Cohort Study. PLoS ONE 2015, 10, e0132826.
- Awad, A.S.; You, H.; Gao, T.; Cooper, T.K.; Nedospasov, S.A.; Vacher, J.; Wilkinson, P.F.; Farrell, F.X.; Reeves, W.B. Macrophage-derived tumor necrosis factor-α mediates diabetic renal injury. Kidney Int. 2015, 88, 722–733.
- Park, M.; Maristany, D.; Huang, D.; Shlipak, M.G.; Whooley, M. Associations of tumor necrosis factor alpha receptor type 1 with kidney function decline, cardiovascular events, and mortality risk in persons with coronary artery disease: Data from the Heart and Soul Study. Atherosclerosis 2017, 263, 68–73.
- Li, G.; Wu, W.; Zhang, X.; Huang, Y.; Wen, Y.; Li, X.; Gao, R. Serum levels of tumor necrosis factor alpha in patients with IgA nephropathy are closely associated with disease severity. BMC Nephrol. 2018, 19, 326.
- Bhagat Singh, A.K.; Jeyaruban, A.S.; Wilson, G.J.; Ranganathan, D. Adalimumab-induced IgA nephropathy. BMJ Case Rep. 2019, 12, e226442.
- Kim, S.K.; Choe, J.Y.; Kwak, S.G.; Bae, J.; Park, S.H.; Lee, H. Effect of tumour necrosis factor-alpha inhibitors on renal function in patients with rheumatoid arthritis from the KOBIO registry from 2012 to 2016. Clin. Exp. Rheumatol. 2018, 36, 1022–1030.
- Mohamed, H.E.; Asker, M.E.; Keshawy, M.M.; Hasan, R.A.; Mahmoud, Y.K. Inhibition of tumor necrosis factor-α enhanced the antifibrotic effect of empagliflozin in an animal model with renal insulin resistance. Mol. Cell. Biochem. 2020, 466, 45–54.
- Youssef, D.M.; El-Shal, A.S.; Hussein, S.; Salah, K.; Ahmed, A. Tumor necrosis factor alpha gene polymorphisms and haplotypes in Egyptian children with nephrotic syndrome. Cytokine 2018, 102, 76–82.
- Ramírez-Bello, J.; Cadena-Sandoval, D.; Mendoza-Rincón, J.F.; Barbosa-Cobos, R.E.; Sánchez-Muñoz, F.; Amezcua-Guerra, L.M.; Sierra-Martínez, M.; Jiménez-Morales, S. Tumor necrosis factor gene polymorphisms are associated with systemic lupus erythematosus susceptibility or lupus nephritis in Mexican patients. Immunol. Res. 2018, 66, 348–354.
- Negishi, H.; Taniguchi, T.; Yanai, H. The Interferon (IFN) Class of Cytokines and the IFN Regulatory Factor (IRF) Transcription Factor Family. Cold Spring Harb. Perspect. Biol. 2018, 10, a028423.
- Anders, H.J.; Lichtnekert, J.; Allam, R. Interferon-alpha and -beta in kidney inflammation. Kidney Int. 2010, 77, 848–854.
- Grzegorzewska, A.E.; Swiderska, M.K.; Warchol, W. Interferon-λ3 as a Predictor of Survival in Hemodialysis Patients. Curr. Mol. Med. 2018, 18, 207–215.
- Liu, S.; Zhang, D.; Zhang, Q.; Liu, W. Interferon-alpha for IgM nephropathy? Clin. Nephrol. 2014, 82, 133–137.
- Zheng, N.; Wang, B.; Fan, J.; Luo, N.; Kong, Q.; Ye, H.; Zhang, J.; Ming, H.; Yu, X. Increased Abundance of Plasmacytoid Dendritic Cells and Interferon-Alpha Induces Plasma Cell Differentiation in Patients of IgA Nephropathy. Mediators Inflamm. 2017, 2017, 4532409.
- Gao, J.; Wei, L.; Liu, X.; Wang, L.; Niu, D.; Jin, T.; Yao, G.; Wang, M.; Yu, Q.; Fu, R. Association Between IFN-γ Gene Polymorphisms and IgA Nephropathy in a Chinese Han Population. Kidney Blood Press. Res. 2017, 42, 136–144.
- Nassar, G.M.; Pedro, P.; Remmers, R.E.; Mohanty, L.B.; Smith, W. Reversible renal failure in a patient with the hypereosinophilia syndrome during therapy with alpha interferon. Am. J. Kidney Dis. 1998, 31, 121–126.
- Ozturk, M.; Basoglu, F.; Yilmaz, M.; Ozagari, A.A.; Baybas, S. Interferon β associated nephropathy in a Multiple Sclerosis patient: A case and review. Mult. Scler. Relat. Disord. 2016, 9, 50–53.
- Migliorini, A.; Angelotti, M.L.; Mulay, S.R.; Kulkarni, O.O.; Demleitner, J.; Dietrich, A.; Sagrinati, C.; Ballerini, L.; Peired, A.; Shankland, S.J.; et al. The antiviral cytokines IFN-α and IFN-β modulate parietal epithelial cells and promote podocyte loss: Implications for IFN toxicity, viral glomerulonephritis, and glomerular regeneration. Am. J. Pathol. 2013, 183, 431–440.
- Poosti, F.; Bansal, R.; Yazdani, S.; Prakash, J.; Post, E.; Klok, P.; van der Born, J.; de Borst, M.H.; van Goor, H.; Poelstra, K.; et al. Selective delivery of IFN-γ to renal interstitial myofibroblasts: A novel strategy for the treatment of renal fibrosis. FASEB J. 2015, 29, 1029–1042.
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873.
- Zhang, Y.; Alexander, P.B.; Wang, X.F. TGF-β Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb. Perspect. Biol. 2017, 9, a022145.
- Lodyga, M.; Hinz, B. TGF-β1—A truly transforming growth factor in fibrosis and immunity. Semin Cell Dev. Biol. 2020, 101, 123–139.
- Sutariya, B.; Jhonsa, D.; Saraf, M.N. TGF-β: The connecting link between nephropathy and fibrosis. Immunopharmacol. Immunotoxicol. 2016, 38, 39–49.
- Isaka, Y. Targeting TGF-β Signaling in Kidney Fibrosis. Int. J. Mol. Sci. 2018, 19, 2532.
- Sun, Y.B.; Qu, X.; Caruana, G.; Li, J. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation 2016, 92, 102–107.
- Higgins, S.P.; Tang, Y.; Higgins, C.E.; Mian, B.; Zhang, W.; Czekay, R.P.; Samarakoon, R.; Conti, D.J.; Higgins, P.J. TGF-β1/p53 signaling in renal fibrogenesis. Cell. Signal. 2018, 43, 1–10.
- Mou, X.; Zhou, D.Y.; Ma, J.R.; Liu, Y.H.; Chen, H.P.; Hu, Y.B.; Shou, C.M.; Chen, J.W.; Liu, W.H.; Ma, G.L. Serum TGF-β1 as a Biomarker for Type 2 Diabetic Nephropathy: A Meta-Analysis of Randomized Controlled Trials. PLoS ONE 2016, 11, e0149513.
- Horikoshi, S.; Fukuda, N.; Tsunemi, A.; Okamura, M.; Otsuki, M.; Endo, M.; Abe, M. Contribution of TGF-β1 and Effects of Gene Silencer Pyrrole-Imidazole Polyamides Targeting TGF-β1 in Diabetic Nephropathy. Molecules 2020, 25, 950.
- Voelker, J.; Berg, P.H.; Sheetz, M.; Duffin, K.; Shen, T.; Moser, B.; Greene, T.; Blumenthal, S.S.; Rychlik, I.; Yagil, Y.; et al. Anti-TGF-β1 Antibody Therapy in Patients with Diabetic Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 953–962.
- Feng, M.; Tang, P.M.; Huang, X.R.; Sun, S.F.; You, Y.K.; Xiao, J.; Lv, L.L.; Xu, A.P.; Lan, H.Y. TGF-β Mediates Renal Fibrosis via the Smad3-Erbb4-IR Long Noncoding RNA Axis. Mol. Ther. 2018, 26, 148–161.
- Wang, P.; Luo, M.L.; Song, E.; Zhou, Z.; Ma, T.; Wang, J.; Jia, N.; Wang, G.; Nie, S.; Liu, Y.; et al. Long noncoding RNA lnc-TSI inhibits renal fibrogenesis by negatively regulating the TGF-β/Smad3 pathway. Sci. Transl. Med. 2018, 10, eaat2039.
- You, Y.K.; Luo, Q.; Wu, W.F.; Zhang, J.J.; Zhu, H.J.; Lao, L.; Lan, H.Y.; Chen, H.Y.; Cheng, Y.X. Petchiether A attenuates obstructive nephropathy by suppressing TGF-β/Smad3 and NF-κB signalling. J. Cell. Mol. Med. 2019, 23, 5576–5587.
- Song, M.K.; Lee, J.H.; Ryoo, I.G.; Lee, S.H.; Ku, S.K.; Kwak, M.K. Bardoxolone ameliorates TGF-β1-associated renal fibrosis through Nrf2/Smad7 elevation. Free Radic. Biol. Med. 2019, 138, 33–42.
- Vinader, V.; Afarinkia, K. A beginner’s guide to chemokines. Future Med. Chem. 2012, 4, 845–852.
- Miller, M.C.; Mayo, K.H. Chemokines from a Structural Perspective. Int. J. Mol. Sci. 2017, 18, 2088.
- Mehta, N.N.; Matthews, G.J.; Krishnamoorthy, P.; Shah, R.; McLaughlin, C.; Patel, P.; Budoff, M.; Chen, J.; Wolman, M.; Go, A.; et al. Higher plasma CXCL12 levels predict incident myocardial infarction and death in chronic kidney disease: Findings from the Chronic Renal Insufficiency Cohort study. Eur. Heart J. 2014, 35, 2115–2122.
- Klimczak-Tomaniak, D.; Pilecki, T.; Żochowska, D.; Sieńko, D.; Janiszewski, M.; Pączek, L.; Kuch, M. CXCL12 in Patients with Chronic Kidney Disease and Healthy Controls: Relationships to Ambulatory 24-Hour Blood Pressure and Echocardiographic Measures. Cardiorenal. Med. 2018, 8, 249–258.
- Chen, L.H.; Advani, S.L.; Thai, K.; Kabir, M.G.; Sood, M.M.; Gibson, I.W.; Yuen, D.A.; Connely, K.A.; Marsden, P.A.; Kelly, D.J.; et al. SDF-1/CXCR4 signaling preserves microvascular integrity and renal function in chronic kidney disease. PLoS ONE 2014, 9, e92227.
- Ribeiro, V.; Bosquetti, B.; Gonçalves, S.M.; Bucharles, S.G.; Rempel, L.; Maciel, R.A.; de Oliveira, S.G.; Pecoits Filho, R.; Marwues Stinghen, A.E. Uremic serum inhibits in vitro expression of chemokine SDF-1: Impact of uremic toxicity on endothelial injury. J. Bras. Nefrol. 2014, 36, 123–131.
- Bouabdallah, J.; Zibara, K.; Issa, H.; Lenglet, G.; Kchour, G.; Caus, T.; Six, I.; Chourkroun, G.; Kamel, S.; Bennis, Y. Endothelial cells exposed to phosphate and indoxyl sulphate promote vascular calcification through interleukin-8 secretion. Nephrol. Dial. Transplant. 2019, 34, 1125–1134.
- Alsharidah, A.S.; Alzogaibi, M.A.; Bayoumy, N.M.; Alghonaim, M. Neutrophil chemokines levels in different stages of nephrotic syndrome. Saudi J. Kidney Dis Transpl. 2017, 28, 1256–1263.
- Lin, Z.; Gong, Q.; Zhou, Z.; Zhang, W.; Liao, S.; Liu, Y.; Yan, X.; Pan, X.; Lin, S.; Li, X. Increased plasma CXCL16 levels in patients with chronic kidney diseases. Eur. J. Clin. Investig. 2011, 41, 836–845.
- Zhao, L.; Wu, F.; Jin, L.; Lu, T.; Yang, L.; Pan, X.; Shao, C.; Li, X.; Lin, Z. Serum CXCL16 as a novel marker of renal injury in type 2 diabetes mellitus. PLoS ONE 2014, 9, e87786.
- Scurt, F.G.; Menne, J.; Brandt, S.; Bernhardt, A.; Mertens, P.R.; Haller, H.; Chatzikyrkou, C.; ROADMAP Steering Committee. Systemic Inflammation Precedes Microalbuminuria in Diabetes. Kidney Int. Rep. 2019, 4, 1373–1386.
- Lee, Y.H.; Kim, K.P.; Park, S.H.; Kim, D.J.; Kim, Y.G.; Moon, J.Y.; Jung, S.W.; Kim, J.S.; Jeong, K.H.; Lee, S.Y.; et al. Urinary chemokine C-X-C motif ligand 16 and endostatin as predictors of tubulointerstitial fibrosis in patients with advanced diabetic kidney disease. Nephrol. Dial. Transplant. 2021, 36, 295–305.
- Tok, A.; Seyithanoğlu, M.; Ozer, A.; Erkayıran, U.; Karaküçük, S.; Çelebi, A. The serum level of soluble CXCL16 is increased in preeclampsia and associated with hepatic/renal damage. J. Matern. Fetal Neonatal Med. 2019, 1–6.
- Abujam, B.; Cheekatla, S.; Aggarwal, A. Urinary CXCL-10/IP-10 and MCP-1 as markers to assess activity of lupus nephritis. Lupus 2013, 22, 614–623.
- Marie, M.A.; Abu Khalil, R.E.; Habib, H.M. Urinary CXCL10: A marker of nephritis in lupus patients. Reumatismo 2014, 65, 292–297.
- Gregg, L.P.; Tio, M.C.; Li, X.; Adams-Huet, B.; de Lemos, J.A.; Hedayati, S.S. Association of Monocyte Chemoattractant Protein-1 with Death and Atherosclerotic Events in Chronic Kidney Disease. Am. J. Nephrol. 2018, 47, 395–405.
- De Oliveira Junior, W.V.; Silva, A.P.F.; de Figueiredo, R.C.; Gomes, K.B.; Simões, E.S.A.C.; Dusse, L.M.S.A.; Rios, D.R. Association between dyslipidemia and CCL2 in patients undergoing hemodialysis. Cytokine 2020, 125, 154858.
- Zhang, W.R.; Craven, T.E.; Malhotra, R.; Cheung, A.K.; Chonchol, M.; Drawz, P.; Sarnak, M.J.; Parikh, C.R.; Shlipak, M.G.; Ix, J.H.; et al. Kidney Damage Biomarkers and Incident Chronic Kidney Disease During Blood Pressure Reduction: A Case-Control Study. Ann. Intern. Med. 2018, 169, 610–618.
- Satirapoj, B. Tubulointerstitial Biomarkers for Diabetic Nephropathy. J. Diabetes Res. 2018, 2018, 2852398.
- Mao, S.; Wu, L. Association between MCP-1 2518 A>G gene polymorphism and chronic kidney disease. Int. Urol. Nephrol. 2018, 50, 2245–2253.
- Vianna, H.R.; Soares, C.M.; Silveira, K.D.; Elmiro, G.S.; Mendes, P.M.; de Sousa Tavares, M.; Teixeira, M.M.; Miranda, D.M.; Silva, A.C.S.E. Cytokines in chronic kidney disease: Potential link of MCP-1 and dyslipidemia in glomerular diseases. Pediatr. Nephrol. 2013, 28, 463–469.
- Musiał, K.; Bargenda, A.; Drożdż, D.; Zwolińska, D. New Markers of Inflammation and Tubular Damage in Children with Chronic Kidney Disease. Dis Markers. 2017, 2017, 9389432.
- Brix, S.R.; Stege, G.; Disteldorf, E.; Hoxha, E.; Krebs, C.; Krohn, S.; Otto, B.; Klätschke, K.; Herden, E.; Heymann, F.; et al. CC Chemokine Ligand 18 in ANCA-Associated Crescentic GN. J. Am. Soc. Nephrol. 2015, 26, 2105–2117.
- Wagrowska-Danilewicz, M.; Danilewicz, M.; Stasikowska, O. CC chemokines and chemokine receptors in IgA nephropathy (IgAN) and in non-IgA mesangial proliferative glomerulonephritis (MesProGN). the immunohistochemical comparative study. Pol. J. Pathol. 2005, 56, 121–126.
- Mansour, S.G.; Puthumana, J.; Coca, S.G.; Gentry, M.; Parikh, C.R. Biomarkers for the detection of renal fibrosis and prediction of renal outcomes: A systematic review. BMC Nephrol. 2017, 18, 72.
- Stroo, I.; Claessen, N.; Teske, G.J.; Butter, L.M.; Florquin, S.; Leemans, J.C. Deficiency for the chemokine monocyte chemoattractant protein-1 aggravates tubular damage after renal ischemia/reperfusion injury. PLoS ONE 2015, 10, e0123203.
- Rudemiller, N.P.; Patel, M.B.; Zhang, J.D.; Jeffs, A.D.; Karlovich, N.S.; Griffiths, R.; Kan, M.J.; Buckley, A.F.; Gunn, M.D.; Crowley, S.D. C-C Motif Chemokine 5 Attenuates Angiotensin II-Dependent Kidney Injury by Limiting Renal Macrophage Infiltration. Am. J. Pathol. 2016, 186, 2846–2856.
- Haller, H.; Bertram, A.; Nadrowitz, F.; Menne, J. Monocyte chemoattractant protein-1 and the kidney. Curr. Opin. Nephrol. Hypertens. 2016, 25, 42–49.
- Zhuang, Q.; Cheng, K.; Ming, Y. CX3CL1/CX3CR1 Axis, as the Therapeutic Potential in Renal Diseases: Friend or Foe? Curr. Gene Ther. 2017, 17, 442–452.
- Fu, D.; Senouthai, S.; Wang, J.; You, Y. FKN Facilitates HK-2 Cell EMT and Tubulointerstitial Lesions via the Wnt/β-Catenin Pathway in a Murine Model of Lupus Nephritis. Front. Immunol. 2019, 10, 784.
- Senouthai, S.; Wang, J.; Fu, D.; You, Y. Fractalkine is Involved in Lipopolysaccharide-Induced Podocyte Injury through the Wnt/β-Catenin Pathway in an Acute Kidney Injury Mouse Model. Inflammation 2019, 42, 1287–1300.
- Luo, R.; Guo, S.M.; Li, Y.Q.; Yang, Y.; Li, M.L.; Han, M.; He, X.F.; Ge, S.W.; Xu, G. Plasma fractalkine levels are associated with renal inflammation and outcomes in immunoglobulin A nephropathy. Nephrol. Dial. Transplant. 2019, 34, 1549–1558.
- You, Y.; Qin, Y.; Lin, X.; Yang, F.; Wang, J.; Yuan, F.; Sooranna, S.R.; Pinhu, L. Upregulated fractalkine levels in Chinese patients with lupus nephritis. Cytokine 2018, 104, 23–28.
- Dong, L.; Nordlohne, J.; Ge, S.; Hertel, B.; Melk, A.; Rong, S.; Haller, H.; von Vietinghoff, S. T Cell CX3CR1 Mediates Excess Atherosclerotic Inflammation in Renal Impairment. J. Am Soc. Nephrol. 2016, 27, 1753–1764.
- Bagci, B.; Bagci, G.; Huzmeli, C.; Sezgin, I.; Ozdemir, O. Associations of fractalkine receptor (CX3CR1) and CCR5 gene variants with hypertension, diabetes and atherosclerosis in chronic renal failure patients undergoing hemodialysis. Int. Urol. Nephrol. 2016, 48, 1163–1170.
- Harjunpää, H.; Llort Asens, M.; Guenther, C.; Fagerholm, S.C. Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front. Immunol. 2019, 10, 1078.
- Khodabandehlou, K.; Masehi-Lano, J.J.; Poon, C.; Wang, J.; Chung, E.J. Targeting cell adhesion molecules with nanoparticles using in vivo and flow-based in vitro models of atherosclerosis. Exp. Biol. Med. 2017, 242, 799–812.
- Chen, J.; Hamm, L.L.; Mohler, E.R.; Hudaihed, A.; Arora, R.; Chen, C.S.; Liu, Y.; Browne, G.; Mills, K.T.; Kleinpeter, M.A.; et al. Interrelationship of Multiple Endothelial Dysfunction Biomarkers with Chronic Kidney Disease. PLoS ONE 2015, 10, e0132047.
- Favretto, G.; Cunha, R.S.D.; Dalboni, M.A.; Oliveira, R.B.; Barreto, F.C.; Massy, Z.A.; Marques Stinghen, A.E. Endothelial Microparticles in Uremia: Biomarkers and Potential Therapeutic Targets. Toxins 2019, 11, 267.
- Bevc, S.; Sabic, S.; Hojs, R. Atherosclerosis in hemodialysis patients--the role of microinflammation. Ren. Fail. 2008, 30, 1012–1016.
- Feng, Y.M.; Thijs, L.; Zhang, Z.Y.; Yang, W.Y.; Huang, Q.F.; Wei, F.F.; Kuznetsova, T.; Jennings, A.M.; Delles, C.; Lennox, R.; et al. Glomerular function in relation to circulating adhesion molecules and inflammation markers in a general population. Nephrol. Dial. Transplant. 2018, 33, 426–435.
- Zhang, Q.; Zhang, M.; Sun, C.; Wang, H.; Tang, T.; Xia, Y.; Shao, Q.; Liu, J.; Jiang, C. Soluble Vascular Cell Adhesion Molecule-1 Is Associated With Disease Activity in Adult-Onset Minimal Change Disease. Am. J. Med. Sci. 2019, 357, 311–315.
- Tycová, I.; Hrubá, P.; Maixnerová, D.; Girmanová, E.; Mrázová, P.; Straňavová, L.; Zachoval, R.; Merta, M.; Slatinská, J.; Kollár, M.; et al. Molecular profiling in IgA nephropathy and focal and segmental glomerulosclerosis. Physiol. Res. 2018, 67, 93–105.
- Lau, Y.C.; Xiong, Q.; Blann, A.D.; Lip, G.Y. Relationship between renal function and circulating microparticles, soluble P-selectin and E-selectin levels in atrial fibrillation. J. Thromb. Thrombolysis. 2017, 43, 18–23.
- Liu, J.J.; Yeoh, L.Y.; Sum, C.F.; Tavintharan, S.; Ng, X.W.; Liu, S.; Lee, S.B.M.; Tang, W.E.; Lim, S.C.; SMART2D Study. Vascular cell adhesion molecule-1, but not intercellular adhesion molecule-1, is associated with diabetic kidney disease in Asians with type 2 diabetes. J. Diabetes Complicat. 2015, 29, 707–712.
- Karimi, Z.; Kahe, F.; Jamil, A.; Marszalek, J.; Ghanbari, A.; Afarideh, M.; Khajeh, E.; Noshad, S.; Esteghamati, A.; Chi, G. Intercellular adhesion molecule-1 in diabetic patients with and without microalbuminuria. Diabetes Metab. Syndr. 2018, 12, 365–368.
- El-Dawla, N.M.Q.; Sallam, A.M.; El-Hefnawy, M.H.; El-Mesallamy, H.O. E-cadherin and periostin in early detection and progression of diabetic nephropathy: Epithelial-to-mesenchymal transition. Clin. Exp. Nephrol. 2019, 23, 1050–1057.
- Xu, Y.; Ling, Y.; Yang, F.; Deng, J.; Rong, L.; Jiang, M.; Jiang, X. The mTOR/p70S6K1 signaling pathway in renal fibrosis of children with immunoglobulin A nephropathy. J. Renin Angiotensin Aldosterone Syst. 2017, 18, 1470320317717831.
- Sawada, K.; Toyoda, M.; Kaneyama, N.; Shiraiwa, S.; Moriya, H.; Miyatake, H.; Tanaka, E.; Yamamoto, N.; Miyauchi, M.; Kimura, M.; et al. Upregulation of α3β1-Integrin in Podocytes in Early-Stage Diabetic Nephropathy. J. Diabetes Res. 2016, 2016, 9265074.
- Zhou, X.; Zhang, J.; Haimbach, R.; Zhu, W.; Mayer-Ezell, R.; Garcia-Calvo, M.; Smith, E.; Price, O.; Kan, Y.; Zycband, E.; et al. An integrin antagonist (MK-0429) decreases proteinuria and renal fibrosis in the ZSF1 rat diabetic nephropathy model. Pharmacol. Res. Perspect. 2017, 5, e00354.