Prion diseases are progressive and transmissive neurodegenerative diseases. The conformational conversion of normal cellular prion protein (PrPC) into abnormal pathogenic prion protein (PrPSc) is critical for its infection and pathogenesis.
- synapse
- amyloid
- calcium homeostasis
- neurotoxicity
1. Introduction
Prion diseases are fatal neurodegenerative diseases, including scrapie in sheep, bovine spongiform encephalopathy in cattle, chronic wasting disease in elk, and Creutzfeldt–Jakob disease (CJD), Gerstmann–Sträussler–Scheinker syndrome, and Kuru in humans. The pathological hallmarks of prion diseases are the spongiform degeneration of glial cells and neurons, synaptic degeneration, and the accumulation of abnormal scrapie-like prion protein (PrPSc) in the brain [1]. It is widely accepted that the conformational conversion of normal cellular prion protein (PrPC) to pathogenic PrPSc is central in the pathogenesis of these diseases. Although PrPC and PrPSc have the same chemical characteristics and primary sequence, PrPSc differs from PrPC in terms of its high content of β-sheet secondary structure, propensity to form insoluble amyloid fibrils, and resistance to protease digestion. When misfolded PrPSc enters the body via the ingestion of contaminated food or iatrogenic contamination, the protease-resistant PrPSc invades the brain, forms aggregates and amyloid fibrils, and in turn promotes neighboring PrPC molecules to misfold and aggregate. Thus, prion diseases are also called transmissible spongiform encephalopathy (TSE).
2. Functions of Normal Cellular Prion Protein and Neurometals
2.1. Prion Protein and Copper
The link between Cu and PrPC was first reported in 1997 [2]. Cu is the third most abundant metal in the brain. It plays vital roles in transmitter synthesis and myelination as a cofactor for numerous enzymes, including cytochrome C, lysyl oxidase, uricase, dopamine hydroxylase, and tyrosinase [3]. Moreover, Cu exhibits neuroprotective activity as a component of Cu/Zn superoxide dismutase (Cu/Zn SOD), which is an endogenous antioxidant. Cu is also implicated in Fe homeostasis as a component of ceruloplasmin, which is a ferroxidase. Recent studies suggest that Cu is stored in synaptic vesicles and is released into the synaptic cleft during neuronal excitation. The released Cu reportedly modulates neuronal activity by binding with NMDA-type glutamate receptors, AMPA-type glutamate receptors, and γ-aminobutyric acid (GABA) receptors [4]. Meanwhile, excess free Cu is toxic because Cu is a redox-active metal that exists as Cu2+ and Cu+ and produces reactive oxygen species (ROS). Orally digested Cu is absorbed from the gastrointestinal pathway through divalent metal transporter 1 (DMT1) and then transported by several transporters such as copper transporter 1 (CTR1) and copper-transporting ATPase (ATP7A and ATP7B). Cu deficiency or excess caused by impairments of these transporters leads to severe neurodegenerative diseases such as Wilson’s disease or Menkes disease [5].
Brown et al. demonstrated that PrP knockout mice exhibited decreased Cu levels in the brain and reduced activity of Cu-dependent enzymes [2]. PrPC is composed of a flexible N-terminal domain and α-helix-rich C-terminal domain that is changed to a β-sheet and involved in the conformational changes of PrPSc (Figure 1).
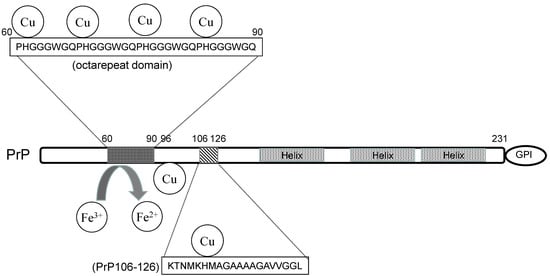
Figure 1. The structure and the metal-binding property of prion protein.
2.2. Prion Protein and Zinc
Other metals including Zn, Fe, and Mn are also associated with prion diseases. Zn2+ has similar chemical characteristics to Cu2+ and shares the same binding proteins; therefore, Zn2+ has the next highest binding affinity to PrPC compared with Cu2+. Zn is the second most abundant trace element in the brain and plays important roles in various physiological functions, such as mitotic cell division, immune system functioning, and synthesis of proteins and DNA, and it acts as a co-factor to more than 300 enzymes and metalloproteins [6]. In the brain, Zn is accumulated in regions such as the cerebral cortex, amygdala, hippocampus, thalamus, and olfactory cortex. Although some Zn firmly binds to metalloproteins or enzymes, a substantial fraction (approximately 10% or more) of Zn either forms free Zn ions (Zn2+) or is loosely bound. Chelatable Zn2+ is stored in the presynaptic vesicles of excitatory glutamatergic neurons and is secreted into the synaptic cleft together with glutamate during neuronal excitation. Synaptic Zn2+ modulates the overall brain excitability by binding to NMDA-type glutamate receptors, GABA receptors, and glycine receptors. Zn2+ also decreases the expression of the GluR2 subunit of AMPA-type glutamate receptors and increases calcium (Ca2+) and/or Zn2+ permeability [7]. Secreted Zn2+ is critical for neuronal communication, synaptic plasticity, and memory formation [8], and therefore, Zn deficiency in children results in dwarfism, delayed mental and physical development, immune dysfunction, and learning disabilities. Zn deficiency also produces learning disorders, taste disorders, and odor disorders in adults [9]. However, excess Zn2+ in pathological conditions such as transient global ischemia causes neuronal death and is central to the pathogenesis of vascular dementia [10].
2.3. Prion Protein and Iron
Fe is the most abundant metal in the brain. Fe is essential for numerous biological functions as an enzyme cofactor for metabolic processes such as oxygen transport, oxidative phosphorylation, and energy transfer. Fe plays critical roles in brain functions such as neurotransmitter synthesis and myelination [11]. Therefore, Fe deficiency impairs learning, especially in children and infants, and it impairs working and learning ability in adults. Fe is a redox active metal and exists in two different forms, ferrous iron (Fe2+) and ferric iron (Fe3+); therefore, excess Fe can generate ROS and is toxic to neurons.
Orally administered Fe is primarily absorbed from the gastrointestinal pathway via DMT-1 as Fe2+. Fe2+ is oxidized to Fe3+ by ferroxidases such as celluroplasmin, and Fe3+ is transported by binding to transferrin. Transferrin-bound Fe3+ passes through the blood–brain barrier and enters into cells via its receptors. Then, Fe3+ is reduced into Fe2+ by ferrireductase, and Fe2+ is transported across membranes by metal transporters and functions as a cofactor for neuronal enzymes. Thus, Fe levels as well as the Fe2+ to Fe3+ ratio are strictly regulated in normal brains.
Increasing evidence suggests that PrPC is involved in Fe homeostasis. Altered Fe metabolism and reduced Fe levels in the brain were observed in PrP knockout mice [12]. Altered ferroxidase and transferrin levels in the cerebrospinal fluid (CSF) of CJD patients have also been reported [13]. PrPC reportedly possesses ferrireductase activity and modulates the cellular uptake of Fe [14]. The octarepeat domain and linkage to the plasma membrane are essential for this activity. Tripath et al. demonstrated that PrPC induces the conversion from Fe3+ to Fe2+, and then Fe2+ is intracellularly transported across membranes by the ZIP14 and DMT-1 complex [15].
It is widely known that the translation of various genes that possess an iron-responsive element (IRE) in their mRNA, such as ferritin or transferrin, is regulated by binding with Fe and iron regulatory proteins (IRPs) [16]. Since the mRNA of the PrPC gene possesses an IRE, the Fe level controls its expression [17].
2.4. Prion Protein and Manganese
Mn is an essential trace element and crucial for various enzymes such as hydrolase, glutamine synthetase, arginase, and pyruvate carboxylase [18]. However, excess Mn is neurotoxic and induces a PD-like syndrome. Mn is absorbed by DMT-1 as well as other divalent cations and is transported by ferroportin, an iron transporter, in addition to Fe2+. Some ZIP transporters (ZIP8 and ZIP14) can transport Mn and Fe.
Mn is suggested to facilitate the pathogenesis of prion diseases [19]. Johnson et al. investigated the levels of trace elements in prion-infected hamster brains using X-ray photoelectron emission microscopy with synchrotron radiation and found reduced Cu and increased Mn in prion protein plaques [20]. Mn enhances the survival of PrP in model soils and increases its infectivity [21]. The risk of chronic wasting disease in elk was associated with a magnesium (Mg) deficiency and increased Mn concentrations [22]. An epidemiological survey in Slovakia suggested a relationship between the pathogenesis of CJD and the imbalance of Mn/Cu in food [23][24]. Moreover, impairment of the Mn transporter is reportedly involved in the infection process [25]. Mn influences Fe homeostasis by affecting the IRE–IRP pathway and causes accumulation of toxic Fe and increased expression of genes with IRE [26].
2.5. Other Amyloidogenic Proteins and Neurometals
Other amyloidogenic proteins such as APP and α-synuclein also possess metal-binding abilities and are involved in the regulation of metal homeostasis. APP possesses two Zn- and/or Cu-binding domains in its N-terminal and the ability to reduce oxidized Cu2+ to Cu+ [27]. Both Zn and Cu are implicated in the dimerization, trafficking, and expression of APP [28]. Cu also affects APP processing and AβP production [29]. Additionally, APP reportedly regulates Fe2+ efflux from cells by binding with ferroportin, which is an Fe2+ transporter [30]. APP mRNA contains an IRE domain similar to ferritin and other Fe-binding proteins [31]. Therefore, APP has endogenous functions in the regulation of the homeostasis of these neurometals and vice versa; thus, these neurometals can control APP expression.
α-Synuclein reportedly binds Cu2+, Mn2+, and other metal ions in its N-terminal and C-terminal domains [32]. In particular, the His50 residue plays a key role in the interaction between Cu and α-synuclein [33]. This His residue may play critical roles in pathogenesis because its mutation is observed in familial-type PD. Metals such as aluminum and Mn enhance the oligomerization of α-synuclein [34]. α-Synuclein possesses ferrireductase activity and converts Fe3+ to Fe2+, similar to PrPC, and it controls neurotransmitter synthesis by providing bioavailable Fe2+ to tyrosine hydroxylase and other enzymes [35]. Indeed, the Fe level and Fe2+ to Fe3+ ratio were reportedly altered in the brains of PD patients [36]. Meanwhile, α-synuclein expression is regulated by Fe levels because its mRNA possesses an IRE domain similar to APP, PrPC, and ferritin [37]. Mn, which has a neurotoxic profile that resembles PD, reportedly induces the overexpression of α-synuclein [38].
2.6. Hypothetical Scheme: Loss of Normal of PrPC Function
Based on these findings, we have developed a hypothetical scheme for the interactions of these amyloidogenic proteins and neurometals at the synapse (Figure 2). During neuronal excitation, Zn2+ and/or Cu2+ are released into the synaptic clefts, and both regulate neuronal excitability by binding to glutamate receptors. The synaptic cleft is a small compartment with a radius of 120 nm and height of 20 nm, and the total volume of the synaptic clefts is estimated to be approximately 1% of the extracellular space of the brain [39]. Thus, it is plausible that Zn2+ and/or Cu2+ levels in the synaptic region may be much higher than those in the CSF. For example, in pathogenic conditions such as transient global ischemia, the Zn2+ concentration reportedly can reach approximately 1~100 µM [40].
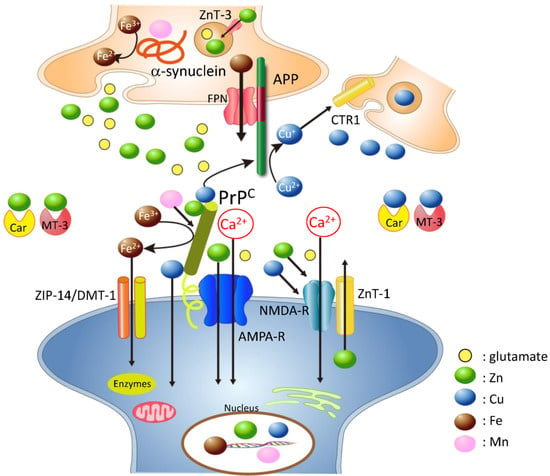
Figure 2. Hypothetical scheme: loss of normal functions of amyloidogenic proteins at the synapse. Normal cellular prion protein (PrPC) is located in the postsynaptic membrane and binds to various receptors. PrPC binds to copper (Cu), zinc (Zn), and iron (Fe) and regulates their levels at the synapse. Additionally, PrPC acts as a ZIP Zn transporter analogue, and the ZnT-1 Zn transporter is also localized at postsynaptic membranes; both proteins control Zn levels at the synapse. PrPC can provide Cu to amyloid precursor protein (APP) or other Cu-binding proteins at the synapse. APP is mainly localized at the presynaptic membrane, binds to Cu and/or Zn, and has the ability to convert Cu2+ to Cu+. APP also regulates Fe2+ efflux from cells via ferroportin. α-Synuclein is mainly localized at the presynaptic domain and binds Cu, manganese (Mn), and Fe. Both PrPC and α-synuclein have ferrireductase activity and provide bioavailable Fe2+ to enzymes at the pre- and postsynaptic regions, respectively. Fe2+ is transported into cells by the ZIP-14 and DMT-1 complex. Other metal-binding factors such as MT-3 and carnosine (Car) are secreted into the synaptic cleft and play critical roles in the maintenance of metal homeostasis. NMDA-R; NMDA-type glutamate receptor, AMPA-R; AMPA-type glutamate receptor, FPN: ferroportin; colored circles represent glutamate, Zn, Cu, Fe, and Mn.
The crosstalk between metals and these amyloidogenic proteins is complex and delicate. When pathogenetic PrPSc enters the brain and causes a depletion of neuroprotective PrPC, the consequent disruption of metal homeostasis will trigger the various adverse effects observed in prion diseases. The loss of PrPC will initiate oxidative damage induced by increased Cu and Fe, increase susceptibility to ROS, deplete neurotransmitters, induce synaptic and neuronal degeneration, and finally cause prion diseases. It is possible that Mn also influences crosstalk by substitution with Cu and by the accumulation of Fe, because Mn affects IRE-IRP binding [26]. It is possible that Mn enhanced neurodegeneration by inducing an overexpression of IRE-containing genes such as α-synuclein, APP, and PrPC. α-Synuclein is reportedly involved in Mn-induced neurodegeneration [41].
This entry is adapted from the peer-reviewed paper 10.3390/ijms22031267
References
- Prusiner, S.B. Biology and genetics of prions causing neurodegeneration. Annu. Rev. Genet. 2013, 47, 601–623.
- Brown, D.R.; Qin, K.; Herms, J.W.; Madlung, A.; Manson, J.; Strome, R.; Fraser, P.E.; Kruck, T.; von Bohlen, A.; Schulz-Schaeffer, W.; et al. The cellular prion protein binds copper in vivo. Nature 1997, 390, 684–687.
- D’Ambrosi, N.; Rossi, L. Copper at synapse: Release, binding and modulation of neurotransmission. Neurochem. Int. 2015, 90, 36–45.
- Gromadzka, G.; Tarnacka, B.; Flaga, A.; Adamczyk, A. Copper Dyshomeostasis in Neurodegenerative Diseases-Therapeutic Implications. Int. J. Mol. Sci. 2020, 21, 9259.
- Kodama, H.; Fujisawa, C.; Bhadhprasit, W. Inherited copper transport disorders: Biochemical mechanisms, diagnosis, and treatment. Curr. Drug Metab. 2012, 13, 237–250.
- Frederickson, C.J.; Suh, S.W.; Silva, D.; Frederickson, C.J.; Thompson, R.B. Importance of zinc in the central nervous system: The zinc-containing neuron. J. Nutr. 2000, 130, 1471S–1483S.
- Calderone, C.; Jover, T.; Mashiko, T.; Noh, K.; Tanaka, H.; Bennett, M.V.L.; Zukin, R. Late calcium EDTA rescues hippocampal CA1 neurons from global ischemia-induced death. J. Neurosci. 2004, 24, 9903–9913.
- Takeda, A.; Tamano, H. The impact of synaptic Zn2+ dynamics on cognition and its decline. Int. J. Mol. Sci. 2017, 18, E2411.
- Prasad, A.S. Impact of the discovery of human zinc deficiency on health. J. Am. Coll. Nutr. 2009, 28, 257–265.
- Kawahara, M.; Mizuno, D.; Koyama, H.; Konoha, K.; Ohkawara, S.; Sadakane, Y. Disruption of zinc homeostasis and the pathogenesis of senile dementia. Metallomics 2014, 6, 209–219.
- Thirupathi, A.; Chang, Y.Z. Brain Iron Metabolism and CNS Diseases. Adv. Exp. Med. Biol. 2019, 1173, 1–19.
- Singh, A.; Kong, Q.; Luo, X.; Petersen, R.B.; Meyerson, H.; Singh, N. Prion protein (PrP) knock-out mice show altered iron metabolism: A functional role for PrP in iron uptake and transport. PLoS ONE 2009, 4, e6115.
- Haldar, S.; Beveridge, J.; Wong, J.; Singh, A.; Galimberti, D.; Borroni, B.; Zhu, X.; Blevins, J.; Greenlee, J.; Perry, G.; et al. A low-molecular-weight ferroxidase is increased in the CSF of sCJD cases: CSF ferroxidase and transferrin as diagnostic biomarkers for sCJD. Antioxid. Redox Signal. 2013, 19, 1662–1675.
- Singh, A.; Haldar, S.; Horback, K.; Tom, C.; Zhou, L.; Meyerson, H.; Singh, N. Prion protein regulates iron transport by functioning as a ferrireductase. J. Alzheimers Dis. 2013, 35, 541–552.
- Tripathi, A.J.; Haldar, S.; Qian, J.; Beserra, A.; Suda, S.; Singh, A.; Hopfer, U.; Chen, S.G.; Garrick, M.D.; Turner, J.R.; et al. Prion protein functions as a ferrireductase partner for ZIP14 and DMT1. Free Radic. Biol. Med. 2015, 84, 322–330.
- Zhou, Z.D.; Tan, E.K. Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 75.
- Rogers, J.T.; Cahill, C.M. Iron-responsive-like elements and neurodegenerative ferroptosis. Learn Mem. 2020, 27, 395–413.
- Tuschl, K.; Mills, P.B.; Clayton, P.T. Manganese and the brain. Int. Rev. Neurobiol. 2013, 110, 277–312.
- Brown, D.R. Prions and manganese: A maddening beast. Metallomics 2011, 3, 229–238.
- Johnson, C.J.; Gilbert, P.; Abrecht, M.; Baldwin, K.L.; Russell, R.E.; Pedersen, J.A.; Aiken, J.M.; McKenzie, D. Low copper and high manganese levels in prion protein plaques. Viruses 2013, 5, 654–662.
- Brown, D.R. Manganese enhances prion protein survival in model soils and increases prion infectivity to cells. PLoS ONE 2009, 4, e7518.
- White, S.N.; O’Rourke, K.I.; Gidlewski, T.; VerCauteren, K.C.; Mousel, M.R.; Phillips, G.E.; Spraker, T.R. Increased risk of chronic wasting disease in Rocky Mountain elk associated with decreased magnesium and increased manganese in brain tissue. Can. J. Vet. Res. 2010, 74, 50–53.
- Slivarichová, D.; Mitrová, E.; Ursínyová, M.; Uhnáková, I.; Koscová, S.; Wsólová, L. Geographic accumulation of Creutzfeldt-Jakob disease in Slovakia--environmental metal imbalance as a possible cofactor. Cent. Eur. J. Public Health 2011, 19, 158–164.
- Masánová, V.; Mitrova, E.; Ursinyova, M.; Uhnakova, I.; Slivarichova, D. Manganese and copper imbalance in the food chain constituents in relation to Creutzfeldt-Jakob disease. Int. J. Environ. Health Res. 2007, 17, 419–428.
- Pass, R.; Frudd, K.; Barnett, J.P.; Blindauer, C.A.; Brown, D.R. Prion infection in cells is abolished by a mutated manganese transporter but shows no relation to zinc. Mol. Cell Neurosci. 2015, 68, 186–193.
- Venkataramani, V.; Doeppner, T.R.; Willkommen, D.; Cahill, C.M.; Xin, Y.; Ye, G.; Liu, Y.; Southon, A.; Aron, A.; Au-Yeung, H.Y.; et al. Manganese causes neurotoxic iron accumulation via translational repression of amyloid precursor protein and H-Ferritin. J. Neurochem. 2018, 147, 831–848.
- Multhaup, G.; Schlicksupp, A.; Hesse, L.; Beher, D.; Ruppert, T.; Masters, C.L.; Beyreuther, K. The amyloid precursor protein of Alzheimer’s disease in the reduction of copper (II) to copper (I). Science 1996, 271, 1406–1409.
- Baumkötter, F.; Schmidt, N.; Vargas, C.; Schilling, S.; Weber, R.; Wagner, K.; Fiedler, S.; Klug, W.; Radzimanowski, J.; Nickolaus, S. Amyloid precursor protein dimerization and synaptogenic function depend on copper binding to the growth factor-like domain. J. Neurosci. 2014, 34, 11159–11172.
- Gerber, H.; Wu, F.; Dimitrov, M.; Garcia Osuna, G.M.; Fraering, P.C. Zinc and copper differentially modulate amyloid precursor protein processing by γ-secretase and amyloid-β peptide production. J. Biol. Chem. 2017, 292, 3751–3767.
- Wong, B.X.; Tsatsanis, A.; Lim, L.Q.; Adlard, P.A.; Bush, A.I.; Duce, J.A. β-Amyloid precursor protein does not possess ferroxidase activity but does stabilize the cell surface ferrous iron exporter ferroportin. PLoS ONE 2014, 9, e114174.
- Rogers, J.T.; Randall, J.D.; Cahill, C.M.; Eder, P.S.; Huang, X.; Gunshin, H.; Leiter, L.; McPhee, J.; Sarang, S.S.; Utsuki, T.; et al. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 2002, 277, 45518–45528.
- Okita, Y.; Rcom-H’cheo-Gauthier, A.N.; Goulding, M.; Chung, R.S.; Faller, P.; Pountney, D.L. Metallothionein, Copper and Alpha-Synuclein in Alpha-Synucleinopathies. Front. Neurosci. 2017, 11, 114.
- Miotto, M.C.; Binolfi, A.; Zweckstetter, M.; Griesinger, C.; Fernández, C.O. Bioinorganic chemistry of synucleinopathies: Deciphering the binding features of Met motifs and His-50 in AS-Cu (I) interactions. J. Inorg. Biochem. 2014, 141, 208–211.
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 2001, 276, 44284–44296.
- Davies, P.; Moualla, D.; Brown, D.R. Alpha-synuclein is a cellular ferrireductase. PLoS ONE 2011, 6, e15814.
- Wang, J.Y.; Zhuang, Q.Q.; Zhu, L.B.; Zhu, H.; Li, T.; Li, R.; Chen, S.F.; Huang, C.P.; Zhang, X.; Zhu, J.H. Meta-analysis of brain iron levels of Parkinson’s disease patients determined by postmortem and MRI measurements. Sci. Rep. 2016, 6, 36669.
- Cahill, C.M.; Lahiri, D.K.; Huang, X.; Rogers, J.T. Amyloid precursor protein and alpha synuclein translation, implications for iron and inflammation in neurodegenerative diseases. Biochim. Biophys. Acta 2009, 1790, 615–628.
- Sun, Y.; He, Y.; Yang, L.; Liang, D.; Shi, W.; Zhu, X.; Jiang, Y.; Ou, C. Manganese induced nervous injury by α-synuclein accumulation via ATP-sensitive K (+) channels and GABA receptors. Toxicol. Lett. 2020, 332, 164–170.
- Schikorski, T.; Stevens, C.F. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J. Neurosci. 1997, 17, 5858–5867.
- Vogt, K.; Mellor, J.; Tong, G.; Nicoll, R. The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron 2000, 26, 187–196.
- Ma, Z.; Liu, K.; Li, X.-R.; Wang, B.; Liu, C.; Yan, D.; Deng, Y.; Liu, W.; Xu, B. Alpha-synuclein is involved in manganese-induced spatial memory and synaptic plasticity impairments via TrkB/Akt/Fyn-mediated phosphorylation of NMDA receptors. Cell Death Dis. 2020, 11, 834.