Cancer is one of the leading causes of death in the world, and antineoplastic drug research continues to be a major field in medicine development. The marine milieu has thousands of biological species that are a valuable source of novel functional proteins and peptides, which have been used in the treatment of many diseases, including cancer. In contrast with proteins and polypeptides, small peptides (with a molecular weight of less than 1000 Da) have overwhelming advantages, such as preferential and fast absorption, which can decrease the burden on human gastrointestinal function. Besides, these peptides are only connected by a few peptide bonds, and their small molecular weight makes it easy to modify and synthesize them. Specifically, small peptides can deliver nutrients and drugs to cells and tissues in the body. These characteristics make them stand out in relation to targeted drug therapy. Nowadays, the anticancer mechanisms of the small marine peptides are still largely not well understood; however, several marine peptides have been applied in preclinical treatment.
- marine organism
- anticancer medicine
- small peptide
- liner peptide
- cyclic peptide
1. Introduction
Oceans cover about 70% of the earth’s surface and 95% of the biosphere. Water was the cradle of the earliest living organisms, containing approximately 75% of all living organisms. The marine environment offers a rich source of natural products with potential therapeutic applications. More than 1 million marine invertebrates and more than 25,000 species of fish have been discovered, and some of these have been shown to contain natural products with potential biological activity [1][2]. In recent years, marine microorganisms, have also been regarded as a valuable source of bioactive compounds, with the advantages of easy cultivation and good compound extraction repeatability [3]. More than 10,000 bioactive molecules that have been isolated from marine organisms, and several have been found to possess anticancer activity [4]. Most of these natural products with anticancer activity originate from microorganisms (bacteria, fungi, protozoa, viruses, and chromista), plantae (flowering plants like mangroves and macroalgae), and animalia (invertebrates such as sponges, tunicates, and vertebrates such as fish and whale), etc.
Cancer is one of the leading causes of death in the world. An estimated 9.6 million people died of cancer in 2018 [5]. Almost 1 in 6 people die of cancer globally. With the application of new theories, modern technologies, and new drugs in basic tumor research and clinical treatment, the rising trend in tumor death in many countries has been effectively controlled [5]. Chemotherapy is part of the major categories of medical oncology. Despite these successes, chemotherapy’s lingering toxic side-effects are still a primary cause of morbidity and mortality in cancer survivors [6]. As for traditional chemotherapy drugs, most of these inhibit tumor cell proliferation by acting on the DNA synthesis and the replication of tumor cells, which have been shown to be effective but at the price of high toxicity due to a lack of selectivity. Nowadays, there are novel molecular methods for treatment using cancer drugs, including target therapy by cell surface receptors, immune-directed therapy, therapeutic vaccines, and antibody–drug conjugates (ADCs) [7][8]. According to the 2020 ASCO’s Annual Report, a large number of innovative drugs, which can be categorized into targeted drugs and immune drugs, have entered trials and clinical trials [9]. Targeted medicine can restrain tumor cell growth by blocking the signal transduction, but the recurrence rate is extremely high [10]. Antibody–drug conjugates have the potential for increased tumor penetration and drug resistance. It has been demonstrated that a knottin peptide–drug conjugate (KDC) can selectively deliver gemcitabine to malignant cells expressing tumor-associated integrins [11]. In recent years, major pharmaceutical companies and research centers have focused on monoclonal antibody drugs and bi-specific antibody drugs in targeted therapy, as well as CAR-T and immunoassay point inhibitors in immunotherapy [12][13][14]. Immunotherapy achieves anti-tumor therapy by stimulating the body’s immune system. In immunotherapy, mainly immune cell therapy, immune checkpoint inhibitors, tumor vaccines, and immune system regulators, the immune system is used to recognize and regulate the body’s attack on abnormal cell functions [14]. Small peptides, such as the thymic peptide, with their unique advantages in immunotherapy, are also very prominent. The thymic peptide used has been a non-specific adjuvant therapy for various tumors, as it can induce T cell differentiation and development, promoting its proliferation and improving T cell response to the antigen at the same time [15]. This kind of drug enhances the patient’s immunity, with fewer side effects.
The resistance adaptation ability of small peptides in many drug treatments and their fewer toxic side effects indicate their potential application in further developing novel drugs. Due to these medicines’ unique metabolic processes, many new study areas on the pharmaceutical aspects of protein and peptide drugs have recently emerged [16][17]. Many natural and synthetic peptides were characterized in recent decades, and public databases were established, such as APD3 (database of antimicrobial peptides), the Defensins Knowledgebase, the antiviral AVPdb (database of antiviral peptides), the antiparasitic ParaPep, and the CancerPPD (database of anticancer peptides and proteins) [18][19][20][21][22]. Bioactive small peptides are composed of 2–10 amino acids linked by peptide bonds. Studies have found that amino acids such as Trp, Tyr, Met, Gly, Cys, His, and Pro in the peptide chain can significantly improve the bioactivity [23][24]. In other words, compared with traditional chemotherapy drugs, small peptides have several advantages, such as high absorption, small size, well-defined signaling targets, and minimal toxicity. As such, they offer a new promising area of research. Besides, small peptides are just connected by a few peptide linkages, and their small molecular weight makes them easy to be modified and synthesized. Additionally, small peptides can be used as vectors to deliver drugs specifically to every cell and tissue [25]. However, small peptides have some disadvantages, including a short half-life, easy degradation in vivo, poor stability, etc. Many researchers have tried to modify or develop corresponding pseudo-peptide drugs or have combined small peptides with traditional therapy for tumor combination therapy to achieve better results than with a single treatment. The application of several cyclic peptides as noncovalent nuclear targeting molecular transporters of Dox has been reported [26]. Additionally, another work designed new peptides based on the molecular dynamics simulation (MDs) of the matuzumab-EGFR complex in a water environment. These peptides had a higher affinity to the EGFR relative to that previously reported [27]. The peptide modification of anticancer drugs could enhance their activity and selectivity, perhaps even circumventing multi-drug resistance [28].
The marine environment is a crucial biological kingdom with the richest source of novel functional proteins and peptides. Additionally, it is gradually becoming a vital field of drug development [29]. As marine organisms live in a special environment that is hypersaline, high-pressure, hypoxia, and hypothermic, and lacks sunlight, these proteins and peptides have strong bioactivities and a specific structure. There are many effects associated with marine peptides, such as antioxidant, antimicrobial, antitumor, antiviral, cardioprotective, immunomodulatory, and tissue regeneration properties [16][30][31][32]. Approximately 49 marine-derived active substances or their derivatives have been approved for the market or entered clinical trials globally [33]. Most of these bioactive molecules are extracted from marine sponges, mollusca, and algae. There are 11 kinds of marine drugs approved by European and American drug authorities, of which, four are listed as anticancer drugs: Cytosar-U, Yondelis, Halaven, and Adcetris. In recent years, more and more studies on marine bioactive peptides have appeared. Many bioactive peptides with anticancer potential have been extracted from various marine organisms. Small peptides from marine sources are gradually gaining attention because of their uniqueness. Compared with high-molecular-weight peptides, low-molecular-weight peptides show greater molecular mobility and diffusivity, contributing to their enhanced interaction with cancer cell components and increasing their anticancer activity [34]. Nowadays, several newly discovered anticancer small peptides and their derivatives thereof from marine organisms have been widely applied to clinical research [35][36][37] (Figure 1). Marine small peptides are molecules that participates in all processes of life activities. They can bear anticancer roles in diversiform aspects in different ways, such as preventing cell migration, induction of apoptosis, disorganization of tubulin structure and inducing cell cycle arrest, and more (Figure 2) [38].

Figure 1. Sources of marine natural products or derivatives that have been approved or entered clinical trials, as well as anticancer small peptides and their derivatives which have entered clinical studies.
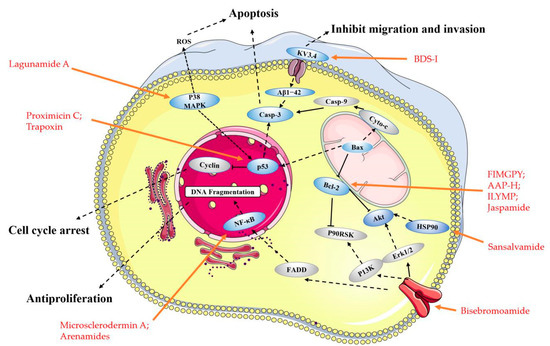
Figure 2. Main molecular mechanism of actions deployed by the anticancer peptides presented in this review. Blue represents the targets mentioned in the paper, and gray represents the proteins involved in the signaling pathway that are not present in this paper, the anticancer peptides mentioned in the article are highlighted in red.
2. Linear Peptides and Derivatives
2.1. Animals
In recent years, some anticancer peptides have been discovered from proteolytic products and secondary metabolites of marine animals, and these purified anticancer peptides have cytotoxic, anti-proliferative and protease inhibition effects [39].
The early discovery of the anti-tumor effect of small marine peptides was mostly based on cytotoxicity assessment, and the subsequent mechanism is not clear. Ding Guo-Fang et al. discovered a tripeptide QPK (Table 1) with anticancer activity shows that it inhibited the growth of DU-145 cells (human prostate cancer cells) in a dose-dependent manner, the IC50 (half-maximal inhibitory concentration) fell from 9.50 mg/mL at 24 h to 1.00 mg/mL at 48 h [40]. Three novel cytotoxic peptides, AGAPGG, AERQ, and RDTQ (Table 1), were successfully purified and identified from the papain hydrolysate of Sarcophyton glaucum. They displayed relatively high cytotoxicity on HeLa cells (human cervical cancer cells), which was 3.3-, 5.8-, and 5.1-fold stronger than that of the anticancer drug 5-FU, respectively. Additionally, their IC50 values to inhibit the growth of Hela cells were 8.6, 4.9, and 5.6 mmol/L, respectively [41].
Table 1. Marine sources of bioactive linear peptides with anticancer potential.
| Compound | Source | Mechanism | Cell Lines | IC50/(GI50) b | Reference |
|---|---|---|---|---|---|
| QPK | Sepia ink | Cytotoxicity a | DU-145 | 9.50 mg/mL (24 h); | [40] |
| 1.00 mg/mL (48 h) | |||||
| AGAPGG; | Sarcophyton glaucum | Cytotoxicity a | HeLa | 8.6 mmol/L; | [41] |
| AERQ; | 4.9 mmol/L; | ||||
| RDTQ | 5.6 mmol/L | ||||
| Virenamides A; | The Didemnid ascidian Diplosoma virens | Inhibiting the Topoisomerase II | P388; | 2.5 µg/mL | [42] |
| A549; | 10 µg/mL | ||||
| HT-29; | 10 µg/mL | ||||
| CV1 | 10 µg/mL | ||||
| Virenamides B | The Didemnid ascidian Diplosoma virens | Inhibiting the Topoisomerase II | P388; | 5 µg/mL | |
| A549; | 5 µg/mL | ||||
| HT-29; | 5 µg/mL | ||||
| CV1 | 5 µg/mL | ||||
| Virenamides C | The Didemnid ascidian Diplosoma virens | Inhibiting the Topoisomerase II | P388; | 5 µg/mL | |
| A549; | 5 µg/mL | ||||
| HT-29; | 5 µg/mL | ||||
| CV1 | 5 µg/mL | ||||
| SCAP1; (Leu-Ala-Asn-Ala-Lys) |
Oyster (Saccostrea cucullata) | Enhancing oxidative DNA damage; Inducing apoptosis |
HT-29 | 90.31 mg/mL (24 h); | [43][44] |
| 70.87 mg/mL (48 h); | |||||
| 60.21 mg/mL (72 h) | |||||
| YALPAH | Half-fin anchovy (Setipinna taty) | Inducing apoptosis | PC‑3 | 8.1 mg/mL | [45][46] |
| BCP-A (Trp-Pro-Pro) |
Blood clam (Tegillarca granosa) muscle | Inducing apoptosis and inhibiting lipid peroxidation | PC-3; | 1.99 mg/mL; | [47] |
| DU-145; | 2.80 mg/mL; | ||||
| H-1299; | 3.3 mg/mL; | ||||
| HeLa | 2.54 mg/mL | ||||
| BDS-I; (Ala-Ala-Pro-Ala-Phe-Ala-Ser-Gly) |
The sea anemone toxin | Blocking KV3.4 currents prevented (the neurotoxic β-amyloid peptide1-42) Aβ1−42-induced caspase-3 activation and apoptotic processes | PC-12 | 75 nM | [48][49] |
| FIMGPY | The skate (R. porosa) cartilage protein hydrolysate | Inducing apoptosis by upregulating the Bax/Bcl-2 ratio and caspase-3 activation | HeLa | 4.81 mg/mL | [50] |
| AAP-H; (Tyr-Val-Pro-Gly-Pro) |
The sea anemone Anthopleura anjunae | Inducing apoptosis, decreasing the mitochondrial membrane potential, and increasing Bax/Bcl-2 ratio, cytochrome-C, caspase-3, and caspase-9 | DU-145 | 9.605 mM (24 h); | [51] |
| 7.910 mM (48 h); | |||||
| 2.298 mM (72 h) | |||||
| ILYMP | Cyclina sinensis | Enhancing expression of Bax, cleaved caspase-3/9 as well as suppression of Bcl-2 expression | DU-145 | 11.25 mM | [52] |
| SCH-P9 (Leu-Pro-Gly-Pro) |
Sinonovacula constricta hydrolysates | Inducing apoptosis and sub-G1 phase cell cycle arrest | DU‑145; | 1.21 mg/mL (24 h); |
[53] |
| PC‑3 | 1.09 mg/mL (24 h) | ||||
| SCH-P10 (Asp-Tyr-Val-Pro) |
DU‑145; | 1.41 mg/mL (24 h); |
|||
| PC‑3 | 0.91 mg/mL (24 h) |
||||
| SIO | Sepia ink | Inducing apoptosis, and S and G2/M phase cell cycle arrest | DU-145; | <5 mg/mL | [54][55] |
| PC-3; | <5 mg/mL | ||||
| LNCaP | <10 mg/mL | ||||
| Psammaplin A (PsA) | The two Sponges, Jaspis sp.and Poecillastra wondoensis. | Inducing S or S-G2/M phase cell cycle arrest; Inhibting HDAC | P388; HCT-116; A549 |
(40 nM) | [56][57] |
| NVP-LAQ824 | Psammaplysilla sp. | Inducing S or S-G2/M phase cell cycle arrest; Inhibting HDAC | H-1299 | 150 nM | [58] |
| HCT-116 | 10 nM | ||||
| Lucentamycins A; | The fermentation broth of a marine-derived actinomycete | Cytotoxicity a | HCT-116 | 0.20 µM; | [59] |
| Lucentamycins B | 11 µM | ||||
| Padanamides A and B | Sediment in the culture of Streptomyces sp. | Cytotoxicity a | Jurkat | 30.9 µM | [60] |
| Tasiamide | Cyanobacterial compound derived from Symploca sp. | Inhibiting the expression of Cath D | KB; | 0.48 μg/mL; | [61][62] |
| LoVo | 3.47 μg/mL | ||||
| Belamide A | Cyanobacterium | Tubulin polymerization inhibition |
HCT-116; | 0.74 μM; | [63] |
| A-10 | 20 μM | ||||
| Symplostatin A | Cyanobacterium | Microtubule assembly Inhibiting cell cycle arrest | MDA-MB-435 | 0.15 μM | [64] |
| SK-OV-3; | 0.09 μM | ||||
| NCI/ADR; | 2.90 μM | ||||
| NCI/ADR with Verapamil; | 0.09 μM | ||||
| A-10; | 1.8 μM | ||||
| HUVEC | 0.16 μM | ||||
| Proximicins C | Actinomycetes of the genus Verrucosispora, | Inducing Cell cycle G1 to S phase arrest and inducing apoptotic cell death | U-87 MG; | 12.7 μg/mL | [65] |
| MDA-MD-231 | 11.4 μg/mL | ||||
| Bisebromoamide | Cyanobacterium of the genus Lyngbya sp. | Inhibiting both the Raf/MEK/ERK and PI3K/Akt/mTOR pathways | JFCR39 | (40 nM) | [66][67] |
| HVLSRAPR | Spirulina platensis | Cytotoxicity a | HT-29; | 99.88 µg/mL | [68] |
Marine natural products are an important source of topological enzyme inhibitors and DNA damaging agents. Virenamides A–C (Table 1 and Figure 3) have been isolated from extracts of the Didemnid ascidian Diplosoma virens. It is reported that Virenamide A exhibited topoisomerase II inhibitory activity and had modest cytotoxicity toward a panel of cultured cells: gave an IC50 of 2.5 μg/mL against P388 (mouse leukemia cells), and 10 μg/mL against A549 (human non-small cells lung cancer cells), HT-29 (human colon cancer cells), and CV1 (kidney cells) cells. Additionally, Virenamides B and C both had an IC50 of 5 μg/mL against P388, A549, HT-29, and CV1 cells [42]. SCAP1 (Table 1) is an anticancer and antioxidative peptide that was shown to initiate cancer cell death by inhibiting cancer cell growth and increasing DNA damage and apoptosis in HT-29 with IC50 values of 90.31 to 60.21 μg/mL [43][44].

Figure 3. The structures of bioactive marine linear peptides and derivatives with anticancer potential.
With the deepening of understanding, researchers have discovered that a variety of marine small peptides can induce tumor cell apoptosis to exert anti-tumor effects. The peptide sequence was identified as YALPAH (Table 1), isolated from half-fin Setipinna taty anchovy, it has been found to induce PC-3 cells (human prostate cancer cells) apoptosis and inhibit cells proliferation, with an IC50 value of 8.1 mg/mL [45]. Additionally, three modified peptides were synthesized ulteriorly. It was revealed that the guanidine portion of arginine (R) forms hydrogen bonds with phosphates, sulfates, and carboxylate salts, which affect the proliferative activity [46]. A tripeptide, BCP-A (Table 1), was isolated from the protein hydrolysate of blood clam (Tegillarca granosa) muscle, showing a strong cytotoxicity toward PC-3, DU-145, H-1299 (human lung cancer cells), and HeLa cells with an IC50 of 1.99, 2.80, 3.3 and 2.54 mg/mL, respectively. Additionally, it was also displaying a high anti-proliferation activity on the PC-3 cells by inducing apoptosis. In addition, BCP-A has a significant anti-lipid peroxidation effect, which is not conducive to tumor formation [47]. A novel peptide obtained from the sea anemone toxin, BDS-I (Table 1), had been successfully identified as a new inhibitor of the KV3.4 channel subunits. In particular, it had been reported that KV3.4 channels play a crucial role in cancer cell migration [69]. BDS-I blocking KV3.4 currents prevented (the neurotoxic β-amyloid peptide1–42) Aβ1−42-induced caspase-3 activation and apoptotic processes [48][49].
Moreover, several small peptides are closely associated with the mitochondrial-mediated apoptosis pathway. The hexapeptide FIMGPY (Table 1), from the skate (Raja porosa) cartilage protein hydrolysate, displayed high anti-proliferation activities in HeLa cells with an IC50 of 4.81 mg/mL. It also could induce apoptosis by upregulating the Bax/Bcl-2 ratio and caspase-3 activation [50]. The anticancer peptide AAP-H (Table 1) is a pentapeptide from the sea anemone Anthopleura anjunae with an amino acid sequence Tyr-Val-Pro-Gly-Pro. It has been shown that AAP-H induces apoptosis by decreasing the mitochondrial membrane potential and increasing Bax/Bcl-2 ratio, cytochrome-C, caspase-3, and caspase-9 [51]. An antiproliferative pentapeptide ILYMP (Table 1), was isolated from the protein hydrolysate of Cyclina sinensis. It has been demonstrated that ILYMP enhances Bax and cleaved caspase-3/9 expression and the suppression of Bcl-2 expression in DU-145 cells [52].
Apoptosis is closely related to cell cycle arrest. At present, some small peptides discovered cannot only induce cancer cells apoptosis, but also cause cell cycle arrest and ultimately lead to cell death. The sequences of SCH-P9 and SCH-P10 (Table 1), identified as Leu-Pro-Gly-Pro and Asp-Tyr-Val-Pro, were obtained from Sinonovacula constricta hydrolysates. The researches illustrated that SCH-P9 and SCH-P10 inhibited the growth of DU-145 cells and PC-3 cells by reducing the number of cells in the G0/G1 phase, thus increasing the number in the sub G1 phase and inducing apoptosis [53]. SIO (Table 1) is another tripeptide found in sepia ink. The research found that it significantly inhibited the proliferation of DU-145, PC-3, LNCaP (human prostate cancer cells), A549 and H-1299 cells, in a time and dose-dependent manner by inducing apoptosis and arresting cell at S or G2/M phase [54][55]. The anticancer mechanism is similar to another decapeptide SHP, which is accompanied by the activation of cellular tumor antigen p53 and caspase-3, the upregulation of pro-apoptosis regulator Bax, and the downregulation of anti-apoptosis regulator Bcl-2 [70]. Psammaplin A (PsA) (Table 1 and Figure 3) is a natural product that has been isolated from sponges and has been suggested to be a promising novel HDAC (histone deacetylase) inhibitor. Some researchers found that PsA exhibited antiproliferative effects on cancer cells by the induction of cell cycle arrest and apoptosis. However, the psammaplin class has the disadvantage of physiologic instability [56][57]. Latest research reports that the indole derivatives of Psammaplin are more potent modulators of epigenetic enzymes than the original natural product. Additionally, positional isomers at the bromoindole ring also showed cell cycle block and apoptosis induction [57]. NVP-LAQ824 (Table 1) is a more stable indolic cinnamyl hydroxamate analogue of Psammaplin A, has entered phase I clinical trials in patients with solid tumors or leukemia [58]. A toxicity evaluation in rats identified the hematopoietic and lymphatic systems as the primary target organs, with a reversible dose-dependent reduction in RBC (red blood cell) and WBC (white blood cell) counts and lymphoid atrophy [58].
2.2. Fungi and Bacteria
Hundreds of secondary metabolites obtained from marine fungal strains revealed potent pharmacological and biological activities [71]. Lucentamycins A–D (Table 1 and Figure 3), have been isolated from the fermentation broth of a marine-derived actinomycete identified by phylogenetic methods as Nocardiopsis lucentensis. Lucentamycins A and B showed significant in vitro cytotoxicity against HCT-116 cells (human colon cancer cells) with IC50 values of 0.20 and 11 µM [59]. Two highly modified linear tetrapeptides, Padanamides A and B (Table 1 and Figure 3), were obtained from sediment in the culture of Streptomyces sp. It demonstrated that Padanamide B is cytotoxic to Jurkat cells (human leukemia cells) with an IC50 value of 30.9 µM [60]. Tasiamide (Table 1 and Figure 3) was predicted to be the best active cyanobacterial compound derived from Symploca sp. It has been shown that it was cytotoxic against KB (human nasopharyngeal cancer cells) and LoVo (human colon cancer cells) cells, with IC50 values of 0.48 and 3.47 μg/mL, respectively [61]. Cathepsin D (Cath D) has been considered a potential target to treat cancer [72]. Tasiamide’s C-terminal modified derivatives have inhibitory activity against Cath D/Cath E/BACE1, potentially making them highly potent and selective Cath D inhibitors [62]. Belamide A (Table 1 and Figure 3) is a highly methylated linear tetrapeptide, with a structural analogy to the important linear peptides, Dolastatins 10 and 15. It has a moderate intensity of cytotoxicity to HCT-116 cells (IC50: 0.74 μM). At a concentration of 20 μM, it destroyed the micro-tubule network in rat aortic smooth muscle A-10 cells and showed the classic tubulin destabilizing mitotic characteristics [63]. Symplostatin A (Table 1 and Figure 3), a Dolastatin 10 analogue from the cyanobacterium Symploca hydnoides, can cause a loss of interphase micro-tubules, G2/M arrest, and active caspase 3 and initiate the phosphorylation of Bcl-2 [64]. Proximicins A, B, and C (Table 1 and Figure 3) are a family of three novel aminofuran antibiotics isolated from actinomycetes of the genus Verrucosispora. It was illustrated that Proximicins could activate cell-cycle regulatory proteins involved in the transition of cells from G1 to S phase and induce apoptotic cell death in L1236 (Hodgkin’s Lymphoma cells) Jurkat 16 (T-cell leukemia cells) cells. Moreover, Proximicin C can induce up-regulation of p53 and p21 in gastric adenocarcinoma cells, and inhibit the U-87 MG (human glioblastoma cells) and MDA-MB-231 (human breast cancer cells) cells proliferation, with IC50 values of 12.7 and 11.4 μg/mL, respectively [65]. Bisebromoamide (Table 1 and Figure 3), a cyanobacterial metabolite from a cyanobacterium of the genus, Lyngbya sp., was shown to have an antiproliferative activity at nanomolar levels of 40 nM that average a 50% growth inhibition (GI50) value across all of the cell lines (a panel of 39 human cancer cell lines (termed JFCR39)) [66]. Additionally, it can also inhibit the Raf/MEK/ERK and PI3K/Akt/mTOR pathways, showing a potent protein kinase inhibitor effect [67].
2.3. Other Small Peptides
Currently, the number of small linear anti-cancer peptides derived from marine plants is relatively small. The peptide, HVLSRAPR (Table 1), exhibited strong inhibitory activity on HT-29 cells, with an IC50 value of 99.88 μg/mL, while it had little inhibitory activity on LO2 cells (normal liver cells) (5.37% at 500 μg/mL). It was also shown to be selectively active on cancer cells, including Hep G2 (human liver cancer cells), MCF-7 (human breast cancer cells), SGC-7901 (human gastric cancer cells), and A549 cells [68].Most of the small peptides are still in the stage of structural optimization and in vitro activity verification. The lack of progress in subsequent research on their mechanisms prevents them from entering clinical development. Additionally, these small peptides merit further investigation as a potential therapeutic agent.
3. Cyclic Peptides and Derivatives
Cyclic peptides combine several favorable properties, such as a good binding affinity, target selectivity, and low toxicity, which make them attractive in anticancer drug researches [73]. Besides, most of the small peptides are concentrated in marine animals, and they are found to be secondary metabolites from sponges, sea squirts, cnidaria, and mollusks.
3.1. Animals
3.1.1. Metabolites of Ascidians
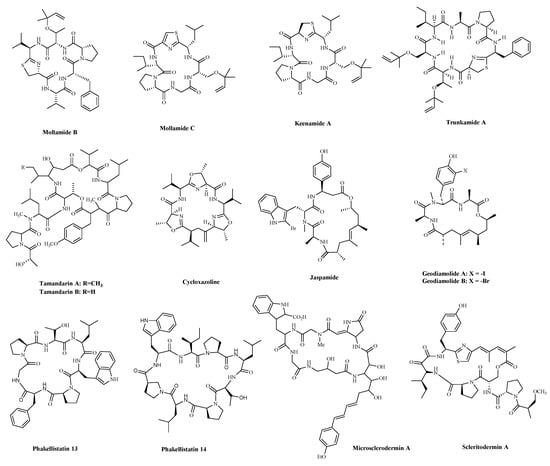
| Compound | Source | Mechanism | Cell Lines | IC50/(GI50) b | Reference |
|---|---|---|---|---|---|
| Mollamide B | Tunicate Didemnum | Cytotoxicity a | H460; | (>100 µM) | [74] |
| MCF-7; | |||||
| SF-268 | |||||
| Keenamide A | Tunicate Didemnum. | Cytotoxicity a | P-388; | 2.5 µg/mL; | [75] |
| A-549; | 2.5 µg/mL; | ||||
| MEL-20; | 2.5 µg/mL; | ||||
| HT-29 | 5.0 µg/mL | ||||
| Trunkamide A | Didemnid ascidians | Cytotoxicity a | DU-145; | 7.08 nM; | [76] |
| IGROV; | 7.31 nM; | ||||
| SK-BR-3; | 5.44 nM; | ||||
| Hela | 3.90 nM | ||||
| Tamandarin A | Didemnid ascidians | Cytotoxicity a | NCI-60 | 1.4 µM (2.3 µM) |
[77] |
| Tamandarin B | NCI-60 | 1.4 µM (2.3 µM) |
|||
| Cycloxazoline | Didemnid ascidians | Cell cycle G2/M arrest, Induction of apoptosis | MRC5CV1; T24 | 0.5 μg/mL | [77] |
| Jaspamide (Jasplakinolide, NSC-613009) |
Sponge Jaspis johnstoni | Induced apoptosis is associated with caspase-3 activation, increased Bax level, and decreased Bcl-2 protein expression | T24; MCF-7; 15NCI/ADR; A-10 |
60 to 150 µg /mL | [79][80] |
| Geodiamolide A | Sponge Geodia corticostylifera | Induction of apoptosis; Tubulin polymerization inhibition |
T47D; | 18.82 nM; | [81] |
| MCF7 | 17.83 nM; | ||||
| Geodiamolides B | T47D; | 113.90 nM; | |||
| MCF7 | 9.82 nM; | ||||
| Phakellistatin 13 | Sponge Phakellia sp. | Induction of both intrinsic and extrinsic apoptosis | BEL-7404 | (10 ng/mL) | [82][83] |
| Phakellistatin 14 | P388 | (5 µg/ mL) | |||
| Microsclerodermin A | Sponge of the genus Amphibleptula | Inhibit NFκB, Induction of apoptosis; | AsPC-1; | 2.3 μM; | [84] |
| BxPC-3; | 0.8 μM; | ||||
| MIA PaCa-2; | 4.3 μM; | ||||
| PANC-1; | 4.0 μM | ||||
| Scleritodermin A | Sponge Scleritoderma nodosum | Tubulin polymerization inhibition |
HCT-116; | 1.9 µM; | [85] |
| A2780; | 0.940 µM; | ||||
| SKBR3 | 0.670 µM |
3.1.2. Metabolites of Sponges
3.2. Fungi

| Compound | Source | Mechanism | Cell Lines | IC50/(GI50) b | Reference |
|---|---|---|---|---|---|
| Zygosporamide | Zygosporium masonii | Cytotoxicity a | SF-268; | (6.5 nM) | [96] |
| RXF 393 | (5.0 nM) | ||||
| Cordyheptapeptide C | Acremonium persicinum | Cytotoxicity a | SF-268; | 3.7 μM; | [97] |
| MCF-7; | 3.0 μM; | ||||
| NCI-H460 | 11.6 μM | ||||
| Cordyheptapeptide D | Acremonium persicinum | Cytotoxicity a | SF-268; | 45.6 μM; | |
| MCF-7; | 82.7 μM; | ||||
| NCI-H460 | >100 μM | ||||
| Cordyheptapeptide E | Acremonium persicinum | Cytotoxicity a | SF-268; | 3.2 μM; | |
| MCF-7; | 2.7 μM; | ||||
| NCI-H460 | 4.5 μM | ||||
| Asperterrestide A | Aspergillus terreus | Cytotoxicity a | U937; | 6.4 μM; | [98] |
| MOLT4 | 6.2 μM | ||||
| Sansalvamide A | Microsporum cf. gypseum | Inhibiting cell growth, and proliferation, and inducing cell apoptosis by regulating the expression of HSP90 | HCT-116; | 1.5 µM; | [99][100] |
| HCT-15 | 1 µM | ||||
| Trapoxin | Fungal product the culture broth of Helicoma ambiens RF-1023 | Inhibiting HDAC | NIH3T3 | 200 ng/mL | [101][102] |
| Microsporin A | Microsporum cf. gypseum | Inhibiting HDAC | HCT-116 | 0.6 mg/mL; | [103] |
| Microsporin B | HCT-116 | 8.5 mg/mL |
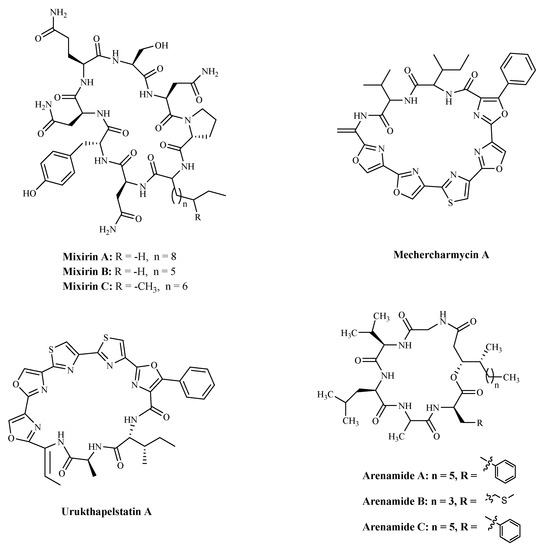
| Compound | Source | Mechanism | Cell Lines | IC50 | Reference |
|---|---|---|---|---|---|
| Mixirin A | Bacillus species. | Cytotoxicity a | HCT-116 | 0.65 µM; | [108] |
| Mixirin B | HCT-116 | 1.6 µM; | |||
| Mixirin C | HCT-116 | 1.26 µM; | |||
| Mechercharmycin A | Thermoactinomyces sp. YM3-251 | Cytotoxicity a | A549; | 40 nM; | [109] |
| Jurkat | 46 nM; | ||||
| Urukthapelstatin A | Thermoactinomycetaceae bacterium Mechercharimyces asporophorigenens YM11-542 | Cytotoxicity a | A549 | 12 nM; | [110] |
| Arenamide A | Salinispora Arenicola. | Inhibiting NF kappa B | 293/NF-κB-Luc | 3.7 μM; | [111] |
| Arenamide B | 293/NF-κB-Luc | 1.7 μM |
This entry is adapted from the peer-reviewed paper 10.3390/md19020115
References
- Molinski, T. Marine natural products. Clin. Adv. Hematol. Oncol. 2009, 7, 383–385.
- de Vries, D.J.; Beart, P.M. Fishing for drugs from the sea: Status and strategies. Trends Pharmacol. Sci. 1995, 16, 275–279.
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2015, 32, 116–211.
- Dyshlovoy, S.A.; Honecker, F. Marine compounds and cancer: 2017 updates. Mar. Drugs 2018, 16, 41.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30.
- Nazish, S.; Mohamed, A. Personalized medicine in cardio-oncology: The role of induced pluripotent stem cell. Cardiovasc. Res. 2019, 115, 949–959.
- Vora, C.; Gupta, S. Targeted therapy in cervical cancer. ESMO Open 2019, 3, e000462.
- Turner, K.A.; Kalafatis, M. The case back on the TRAIL: Death receptors as markers for rhTRAIL sensitivity. J. Appl. Lab. Med. 2017, 2, 176–185.
- Salazar, R.; Cortés-Funes, H.; Casado, E.; Pardo, B.; López-Martín, A.; Cuadra, C.; Tabernero, J.; Coronado, C.; García, M.; Soto Matos-Pita, A.; et al. Phase I study of weekly kahalalide F as prolonged infusion in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 72, 75–83.
- Jiao, S.; Wang, H.; Shi, Z.; Dong, A.; Zhang, W.; Song, X.; He, F.; Wang, Y.; Zhang, Z.; Wang, W.; et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 2014, 25, 166–180.
- Cox, N.; Kintzing, J.R.; Smith, M.; Grant, G.A.; Cochran, J.R. Integrin-targeting knottin peptide-drug conjugates are potent inhibitors of tumor cell proliferation. Angew. Chem. Int. Ed. Engl. 2016, 55, 9894–9897.
- Bjork, R.L. Bi-Specific Monoclonal Antibody (Specific for Both CD3 and CD11b) Therapeutic Drug. U.S. Patent WO2007US14524, 22 June 2007.
- Isazadeh, A.; Hajazimian, S.; Garshasbi, H.; Shadman, B.; Baradaran, B. Resistance mechanisms to immune checkpoints blockade by monoclonal antibody drugs in cancer immunotherapy: Focus on myeloma. J. Cell. Physiol. 2020, 236, 791–805.
- Shahidian, A.; Ghassemi, M.; Mohammadi, J.; Hashemi, M. Immunotherapy. In Bio-Engineering Approaches to Cancer Diagnosis and Treatment; Academic Press: Cambridge, MA, USA, 2020; pp. 69–114.
- Lian, Q.; Cheng, Y.; Zhong, C.; Wang, F. Inhibition of the IgE-mediated activation of RBL-2H3 cells by TIPP, a novel thymic immunosuppressive pentapeptide. Int. J. Mol. Sci. 2015, 16, 2252–2268.
- Patil, S.; Vhora, I.; Amrutiya, J.; Lalani, R.; Misra, A. Role of nanotechnology in delivery of protein and peptide drugs. Curr. Pharm. Des. 2015, 21, 4155–4173.
- Tatiana, R.; Andreas, B.; Yves, S.; Christopher, K.; Alison, B.; Fabien, F.; Luca, M.; Ismael, Z.; Andrea, M. Software-aided approach to investigate peptide structure and metabolic susceptibility of amide bonds in peptide drugs based on high resolution mass spectrometry. PLoS ONE 2017, 12, e0186461.
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093.
- Seebah, S.; Suresh, A.; Zhuo, S.; Choong, Y.H.; Chua, H.; Chuon, D.; Beuerman, R.; Verma, C. Defensins knowledgebase: A manually curated database and information source focused on the defensins family of antimicrobial peptides. Nucleic Acids Res. 2007, 35, D265–D268.
- Qureshi, A.; Thakur, N.; Tandon, H.; Kumar, M. AVPdb: A database of experimentally validated antiviral peptides targeting medically important viruses. Nucleic Acids Res. 2014, 42, D1147–D1153.
- Divya, M.; Priya, A.; Vineet, K.; Anshika, J.; Deepika, M.; Sandeep, S.; Abhishek, T.; Kumardeep, C.; Gautam, S.K.; Ankur, G. ParaPep: A web resource for experimentally validated antiparasitic peptide sequences and their structures. Database (Oxf.) 2014.
- Atul, T.; Abhishek, T.; Priya, A.; Sudheer, G.; Minakshi, S.; Deepika, M.; Anshika, J.; Sandeep, S.; Ankur, G.; Raghava, G.P.S. CancerPPD: A database of anticancer peptides and proteins. Nucleic Acids Res. 2015, 43, 837–843.
- Zhang, W.J.; Wang, S.; Kang, C.Z.; Lv, C.G.; Zhou, L.; Huang, L.Q.; Guo, L.P. Pharmacodynamic material basis of traditional chinese medicine based on biomacromolecules: A review. Plant Methods 2020, 16, 26.
- Chalamaiah, M.; Yu, W.; Wu, J. Immunomodulatory and anticancer protein hydrolysates (peptides) from food proteins: A review. Food Chem. 2018, 245, 205–222.
- Blanco-Míguez, A.; Gutiérrez-Jácome, A.; Pérez-Pérez, M.; Pérez-Rodríguez, G.; Catalán-García, S.; Fdez-Riverola, F.; Lourenço, A.; Sánchez, B. From amino acid sequence to bioactivity: The biomedical potential of antitumor peptides. Protein Sci. 2016, 25, 1084–1095.
- Nasrolahi Shirazi, A.; Tiwari, R.; Chhikara, B.S.; Mandal, D.; Parang, K. Design and biological evaluation of cell-penetrating peptide-doxorubicin conjugates as prodrugs. Mol. Pharm. 2013, 10, 488–499.
- Ebrahimi, M.; Mani-Varnosfaderani, A.; Khayamian, T.; Gharaghani, S. An in silico approach to design peptide mimetics based on docking and molecular dynamics simulation of EGFR-matuzumab complex. J. Iran Chem. Soc. 2016, 13, 1805–1817.
- Gellerman, G.; Baskin, S.; Galia, L.; Gilad, Y.; Firer, M.A. Drug resistance to chlorambucil in murine B-cell leukemic cells is overcome by its conjugation to a targeting peptide. Anticancer Drugs 2013, 24, 112–119.
- Khalifa, S.A.M.; Elias, N.; Farag, M.A.; Chen, L.; Saeed, A.; Hegazy, M.F.; Moustafa, M.S.; Abd El-Wahed, A.; Al-Mousawi, S.M.; Musharraf, S.G.; et al. Marine natural products: A source of novel anticancer drugs. Mar. Drugs 2019, 17, 491.
- Ganesan, A.R.; Mohanram, M.S.G.; Balasubramanian, B.; Kim, I.H.; Seedevi, P.; Mohan, K.; Kanagasabai, S.; Arasu, M.V.; Al-Dhabi, N.A.; Ignacimuthu, S. Marine invertebrates’ proteins: A recent update on functional property. J. King Saud Univ. Sci. 2020, 32, 1496–1502.
- Cheung, R.C.; Ng, T.B.; Wong, J.H. Marine peptides: Bioactivities and applications. Mar. Drugs 2015, 13, 4006–4043.
- Narayanasamy, A.; Balde, A.; Raghavender, P.; Shashanth, D.; Abraham, J.; Joshi, I.; Nazeer, R.A. Isolation of marine crab (Charybdis natator) leg muscle peptide and its anti-inflammatory effects on macrophage cells. Biocatal. Agric. Biotechnol. 2020, 25, 101577.
- Wang, C.; Zhang, G.J.; Liu, W.D.; Yang, X.Y.; Zhu, N.; Shen, J.M.; Wang, Z.C.; Liu, Y.; Cheng, S.; Yu, G.L.; et al. Recent progress in research and development of marine drugs. Chin. J. Mar. Drugs 2019, 38, 35–69.
- Kim, S.M. Antioxidant and anticancer activities of enzymatic hydrolysates of solitary tunicate (Styela clava). Food Sci. Biotechnol. 2011, 20, 1075–1085.
- Shakeel, E.; Arora, D.; Jamal, Q.M.S.; Akhtar, S.; Khan, M.K.A.; Kamal, M.A.; Siddiqui, M.H.; Lohani, M.; Arif, J.M. Marine drugs: A hidden wealth and a new epoch for cancer management. Curr. Drug Metab. 2018, 19, 523–543.
- Dyshlovoy, S.; Honecker, F. Marine compounds and cancer: Updates 2020. Mar. Drugs 2020, 18, 643.
- Dyshlovoy, S.; Honecker, F. Marine compounds and cancer: The first two decades of XXI century. Mar. Drugs 2019, 18, 20.
- Srinivasan, N.; Dhanalakshmi, S.; Pandian, P. Encouraging leads from marine sources for cancer therapy—A review approach. Pharmacogn. J. 2020, 12, 1475–1481.
- Ming-Jia, Z.; Dong-Mei, Z.; Jun-Hui, C. Research advances of antitumor peptides. Chin. J. Biochem. Pharm. 2007, 28, 139–141.
- Ding, G.-F.; Huang, F.-F.; Yang, Z.-S.; Yu, D.; Yang, Y.-F. Anticancer activity of an oligopeptide isolated from hydrolysates of Sepia Ink. Chin. J. Nat. Med. 2011, 9, 151–155.
- Quah, Y. Purification and identification of novel cytotoxic oligopeptides from soft coral Sarcophyton glaucum. J. Zhejiang Univ. Sci. B Biomed. Biotechnol. 2019, 20, 59–70.
- Gan, H. Concise and efficient total syntheses of virenamides A and D. J. Adv. Chem. 2008, 4, 488–493.
- Umayaparvathi, S.; Arumugam, M.; Meenakshi, S.; Dräger, G.; Kirschning, A.; Balasubramanian, T. Purification and characterization of antioxidant peptides from oyster (Saccostrea cucullata) hydrolysate and the anticancer activity of hydrolysate on human colon cancer cell lines. Int. J. Pept. Res. Ther. 2014, 20, 231–243.
- Umayaparvathi, S.; Meenakshi, S.; Vimalraj, V.; Arumugam, M.; Sivagami, G.; Balasubramanian, T. Antioxidant activity and anticancer effect of bioactive peptide from enzymatic hydrolysate of oyster (Saccostrea cucullata). Biomed. Prev. Nutr. 2014, 4, 343–353.
- Song, R.; Wei, R.B.; Luo, H.Y.; Yang, Z.S. Isolation and identification of an antiproliferative peptide derived from heated products of peptic hydrolysates of half-fin anchovy (Setipinna taty). J. Funct. Foods 2014, 10, 104–111.
- Ratih, P.; Se-Kwon, K. Bioactive peptide of marine origin for the prevention and treatment of non-communicable diseases. Mar. Drugs 2017, 15, 67.
- Chi, C.-F.; Hu, F.-Y.; Wang, B.; Li, T.; Ding, G.-F. Antioxidant and anticancer peptides from the protein hydrolysate of blood clam (Tegillarca granosa) muscle. J. Funct. Foods 2015, 15, 301–313.
- Ciccone, R.; Piccialli, I.; Grieco, P.; Merlino, F.; Annunziato, L.; Pannaccione, A. Synthesis and pharmacological evaluation of a novel peptide based on anemonia sulcata BDS-I toxin as a new KV3.4 inhibitor exerting a neuroprotective effect against amyloid-β peptide. Front. Chem. 2019, 9, 497.
- Min, S.; Su, P.; Jeong, P.; Jin, B.; Hee, J.; Seung, S.; Pan, R.; So, L. Kv3.1 and Kv3.4, are involved in cancer cell migration and invasion. Int. J. Mol. Sci. 2018, 19, 1061.
- Pan, X.; Zhao, Y.Q.; Hu, F.Y.; Chi, C.F.; Wang, B. Anticancer activity of a hexapeptide from skate (Raja porosa) cartilage protein hydrolysate in HeLa cells. Mar. Drugs 2016, 14, 153.
- Wu, Z.Z.; Ding, G.F.; Huang, F.F.; Yang, Z.S.; Yu, F.M.; Tang, Y.P.; Jia, Y.L.; Zheng, Y.Y.; Chen, R. Anticancer activity of anthopleura anjunae oligopeptides in prostate cancer DU-145 cells. Mar. Drugs 2018, 16, 125.
- Fangmiao, Y.; Yaru, Z.; Lei, Y.; Yunping, T.; Guofang, D.; Xiaojun, Z.; Zuisu, Y. A novel antiproliferative pentapeptide (ILYMP) isolated from cyclinasinensis protein hydrolysate induces apoptosis of DU145 prostate cancer cells. Mol. Med. Rep. 2018, 18, 771–778.
- Huang, F.; Ding, G.; Yang, Z.; Yu, F. Two novel peptides derived from Sinonovacula constricta inhibit the proliferation and induce apoptosis of human prostate cancer cells. Mol. Med. Rep. 2017, 16, 6697–6707.
- Zhang, Z.; Sun, L.; Zhou, G.; Xie, P.; Ye, J. Sepia ink oligopeptide induces apoptosis and growth inhibition in human lung cancer cells. Oncotarget 2017, 8, 23202–23212.
- Huang, F.; Yang, Z.; Yu, D.; Wang, J.; Li, R.; Ding, G. Sepia ink oligopeptide induces apoptosis in prostate cancer cell lines via caspase-3 activation and elevation of Bax/Bcl-2 ratio. Mar Drugs 2012, 10, 2153–2165.
- Baud, M.G.J.; Leiser, T.; Haus, P.; Samlal, S.; Wong, A.C.; Wood, R.J.; Petrucci, V.; Gunaratnam, M.; Hughes, S.M.; Buluwela, L. Defining the mechanism of action and enzymatic selectivity of Psammaplin A against its epigenetic targets. J. Med. Chem. 2012, 55, 1731–1750.
- Kumar M, S.L.; Ali, K.; Chaturvedi, P.; Meena, S.; Datta, D.; Panda, G. Design, synthesis and biological evaluation of oxime lacking Psammaplin inspired chemical libraries as anti-cancer agents. J. Mol. Struct. 2021, 1225.
- Simmons, T.L.; Andrianasolo, E.; McPhail, K.; Flatt, P.; Gerwick, W.H. Marine natural products as anticancer drugs. Mol. Cancer Ther. 2005, 4, 333–342.
- Cho, J.Y.; Williams, P.G.; Kwon, H.C.; Jensen, P.R.; Fenical, W. Lucentamycins A–D, cytotoxic peptides from the marine-derived actinomycete nocardiopsis lucentensis. J. Nat. Prod. 2007, 70, 1321–1328.
- Williams, D.E.; Dalisay, D.S.; Patrick, B.O.; Matainaho, T.; Andrusiak, K.; Deshpande, R.; Myers, C.L.; Piotrowski, J.S.; Boone, C.; Yoshida, M.; et al. Padanamides A and B, highly modified linear tetrapeptides produced in culture by a Streptomyces sp. isolated from a marine sediment. Org. Lett. 2011, 13, 3936–3939.
- Williams, P.G.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Tasiamide, a cytotoxic peptide from the marine cyanobacterium Symploca sp. J. Nat. Prod. 2002, 65, 1336.
- Li, Z.; Bao, K.; Xu, H.; Wu, P.; Li, W.; Liu, J.; Zhang, W. Design, synthesis, and bioactivities of tasiamide B derivatives as cathepsin D inhibitors. J. Pept. Sci. 2019, 25, e3154.
- Simmons, T.L.; Mcphail, K.L.; Ortega-Barría, E.; Mooberry, S.L.; Gerwick, W.H. Belamide A, a new antimitotic tetrapeptide from a Panamanian marine cyanobacterium. Tetrahedron Lett. 2006, 37, 3387–3390.
- Corbett, T.H. The molecular pharmacology of symplostatin 1: A new antimitotic dolastatin 10 analog. Int. J. Oncol. 2010, 104, 512–521.
- Brucoli, F.; Natoli, A.; Marimuthu, P.; Borrello, M.T.; Stapleton, P.; Gibbons, S.; Schätzlein, A. Efficient synthesis and biological evaluation of proximicins A, B and C. Bioorg. Med. Chem. 2012, 20, 2019–2024.
- Teruya, T.; Sasaki, H.; Fukazawa, H.; Suenaga, K. Bisebromoamide, a potent cytotoxic peptide from the marine cyanobacterium Lyngbya sp.: Isolation, stereostructure, and biological activity. Org. Lett. 2010, 41, 5062–5065.
- Suzuki, K.; Mizuno, R.; Suenaga, K.; Teruya, T.; Tanaka, N.; Kosaka, T.; Oya, M. Bisebromoamide, an extract from Lyngbya species, induces apoptosis through ERK and mTOR inhibitions in renal cancer cells. Cancer Med. 2013, 2, 32–39.
- Wang, Z.; Zhang, X. Isolation and identification of anti-proliferative peptides from Spirulina platensis using three-step hydrolysis. J. Sci. Food Agric. 2017, 97, 918–922.
- Aissaoui, D.; Mlayah Bellalouna, S.; Jebali, J.; Abdelkafi Koubaa, Z.; Souid, S.; Moslah, W.; Othman, H.; Luis, J.; Elayeb, M.; Marrakchi, N. Functional role of Kv1.1 and Kv1.3 channels in the neoplastic progression steps of three cancer cell lines, elucidated by scorpion peptides. Int. J. Biol. Macromol. 2018, 111, 1146–1155.
- Huang, F.; Jing, Y.; Ding, G.; Yang, Z. Isolation and purification of novel peptides derived from Sepia ink: Effects on apoptosis of prostate cancer cell PC-3. Mol. Med. Rep. 2017, 16, 4222–4228.
- Youssef, F.S.; Ashour, M.L.; Singab, A.N.B.; Wink, M. A Comprehensive review of bioactive peptides from marine fungi and their biological significance. Mar. Drugs 2019, 17, 559.
- Pranjol, M.Z.I.; Gutowski, N.J.; Hannemann, M.; Whatmore, J.L. Cathepsin D non-proteolytically induces proliferation and migration in human omental microvascular endothelial cells via activation of the ERK1/2 and PI3K/AKT pathways. Biochim. Biophys. Acta. Mol. Cell. Res. 2018, 1865, 25–33.
- Choi, J.S.; Joo, S.H. Recent trends in cyclic peptides as therapeutic agents and biochemical tools. Biomol. Ther. 2020, 28, 18–24.
- Donia, M.S.; Wang, B.; Dunbar, D.C.; Desai, P.V.; Hamann, M.T. Mollamides B and C, cyclic hexapeptides from the indonesian tunicate Didemnum molle. Planta Med. 2008, 71, 941–945.
- Wesson, K.J.; Hamann, M.T. Keenamide A, a bioactive cyclic peptide from the marine mollusk Pleurobranchus forskalii. J. Nat. Prod. 1996, 59, 629–631.
- Mckeever, B.; Pattenden, G. Total synthesis of trunkamide A, a novel thiazoline-based prenylated cyclopeptide metabolite from Lissoclinum sp. Tetrahedron 2003, 59, 2713–2727.
- Liang, B.; Richard, D.J.; Portonovo, P.S.; Joullié, M.M. Total syntheses and biological investigations of tamandarins A and B and tamandarin A analogs. J. Am. Chem. Soc. 2001, 123, 4469–4474.
- Watters, D.J.; Beamish, H.J. Accumulation of HL-60 leukemia cells in G2/M and inhibition of cytokinesis caused by two marine compounds, bistratene A and cycloxazoline. Cancer Chemoth. Pharm. 1994, 33, 399–409.
- Schweikart, K.; Guo, L.; Shuler, Z.; Abrams, R.; Chiao, E.T.; Kolaja, K.L.; Davis, M. The effects of jaspamide on human cardiomyocyte function and cardiac ion channel activity. Toxicol. In Vitro 2013, 27, 745–751.
- Cioca, D.P.; Kitano, K. Induction of apoptosis and CD10/neutral endopeptidase expression by jaspamide in HL-60 line cells. Cell Mol. Life Sci. 2002, 59, 1377–1387.
- Rangel, M.; Prado, M.P.; Konno, K.; Naoki, H.; Freitas, J.C.; Machado-Santelli, G.M. Cytoskeleton alterations induced by Geodia corticostylifera depsipeptides in breast cancer cells. Peptides 2006, 27, 2047–2057.
- Meli, A.; Tedesco, C.; Della Sala, G.D.; Schettini, R.; Albericio, F.; De Riccardis, F.; Izzo, I. Phakellistatins: An underwater unsolved puzzle. Mar. Drugs 2017, 15, 78.
- Pettit, G.R.; Tan, R. Isolation and structure of phakellistatin 14 from the western pacific marine sponge Phakellia sp. J. Nat. Prod. 2005, 68, 60–63.
- Guzmán, E.A.; Maers, K.; Roberts, J.; Kemami-Wangun, H.V.; Harmody, D.; Wright, A.E. The marine natural product microsclerodermin A is a novel inhibitor of the nuclear factor kappa B and induces apoptosis in pancreatic cancer cells. Investig. New Drugs. 2015, 33, 86.
- Schmidt, E.W.; Raventos Suarez, C.; Bifano, M.; Menendez, A.T.; Fairchild, C.R.; Faulkner, D.J. Scleritodermin A, a cytotoxic cyclic peptide from the lithistid sponge scleritoderma nodosum. J. Nat. Prod. 2004, 67, 475–478.
- Robinson, S.J.; Morinaka, B.I.; Amagata, T.; Tenney, K.; Crews, P. New structures and bioactivity properties of jasplakinolide (jaspamide) analogues from marine sponges. J. Med. Chem. 2010, 53, 1651–1661.
- Rangel, M.; Ionta, M.; Pfister, C.S.; Ferreira, A.S.A.R.; Machado-Santelli, M.G. Marine sponge depsipeptide increases gap junction length in HTC cells transfected with Cx43–GFP. Cell Biol. Int. Rep. 2013, 17, 13–18.
- Pettit, G.R.; Cichacz, Z.; Barkoczy, J.; Dorsaz, A.C.; Herald, D.L.; Williams, M.D.; Doubek, D.L.; Schmidt, J.M.; Tackett, L.P.; Brune, D.C. Isolation and structure of the marine sponge cell growth inhibitory cyclic peptide phakellistatin 1. J. Nat. Prod. 1993, 56, 260–267.
- Wang, S.; Crevenna, A.H.; Ugur, I.; Marion, A.; Antes, I.; Kazmaier, U.; Hoyer, M.; Lamb, D.C.; Gegenfurtner, F.; Kliesmete, Z.; et al. Actin stabilizing compounds show specific biological effects due to their binding mode. Sci. Rep. 2019, 9, 9731.
- Chan, W.R.; Tinto, W.F.; Manchand, P.S.; Todaro, L.J. Stereostructures of Geodiamolides A and B, novel cyclodepsipeptides from the marine sponge Geodia sp. J. Org. Chem. 1987, 52, 3091–3093.
- Tinto, W.F.; Lough, A.J.; McLean, S.; Reynolds, W.F.; Yu, M.; Chan, W.R. Geodiamolides H and I, further cyclodepsipeptides from the marine sponge Geodia sp. Tetrahedron 1998, 54, 4451–4458.
- Freitas, V.M.; Rangel, M.; Bisson, L.F.; Jaeger, R.G.; Machado-Santelli, G.M. The geodiamolide H, derived from Brazilian sponge Geodia corticostylifera, regulates actin cytoskeleton, migration and invasion of breast cancer cells cultured in three-dimensional environment. J. Cell. Physiol. 2008, 216, 583–594.
- Bao, Y.; Jiang, S.; Zhao, L.; Jin, Y.; Yan, R.; Wang, Z. Photoinduced synthesis and antitumor activity of a phakellistatin 18 analog with an isoindolinone fragment. New J. Chem. 2020, 44, 19174–19178.
- Tian, T.; Takada, K.; Ise, Y.; Ohtsuka, S.; Okada, S.; Matsunaga, S. Microsclerodermins N and O, cytotoxic cyclic peptides containing a p-ethoxyphenyl moiety from a deep-sea marine sponge Pachastrella sp. Tetrahedron 2020, 76, 130997.
- Vinothkumar, S.; Parameswaran, P.S. Recent advances in marine drug research. Biotechnol. Adv. 2013, 31, 1826–1845.
- Oh, D.C.; Jensen, P.R.; Fenical, W. Zygosporamide, a cytotoxic cyclic depsipeptide from the marine-derived fungus Zygosporium masonii. Tetrahedron Lett. 2006, 47, 8625–8628.
- Chen, Z.; Song, Y.; Chen, Y.; Huang, H.; Zhang, W.; Ju, J. Cyclic heptapeptides, cordyheptapeptides C–E, from the marine-derived fungus Acremonium persicinum SCSIO 115 and their cytotoxic activities. J. Nat. Prod. 2012, 75, 1215–1219.
- He, F.; Bao, J.; Zhang, X.Y.; Tu, Z.C.; Shi, Y.M.; Qi, S.H. Asperterrestide A, a cytotoxic cyclic tetrapeptide from the marine-derived fungus Aspergillus terreus SCSGAF0162. J. Nat. Prod. 2013, 76, 1182–1186.
- Ujiki, M.B.; Milam, B.; Ding, X.Z.; Roginsky, A.B.; Salabat, M.R.; Talamonti, M.S.; Bell, R.H.; Gu, W.; Silverman, R.B.; Adrian, T.E. A novel peptide sansalvamide analogue inhibits pancreatic cancer cell growth through G0/G1 cell-cycle arrest. Biochem. Biophys. Res. Commun. 2006, 340, 1224–1228.
- Wang, X.; Zhang, J.; Wu, H.; Li, Y.; Conti, P.S.; Chen, K. PET imaging of Hsp90 expression in pancreatic cancer using a new 64 Cu-labeled dimeric Sansalvamide A decapeptide. Amino Acids 2018, 50, 897–907.
- Kijima, M.; Yoshida, M.; Sugita, K.; Horinouchi, S.; Beppu, T. Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J. Biol. Chem. 1993, 268, 22429–22435.
- Jung, M.; Hoffmann, K.; Brosch, G.; Loidl, P. Analogues of trichostatin A and trapoxin B as histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 1997, 7, 1655–1658.
- Gu, W.; Cueto, M.; Jensen, P.R.; Fenical, W.; Silverman, R.B. Microsporins A and B: New histone deacetylase inhibitors from the marine-derived fungus Microsporum cf. gypseum and the solid-phase synthesis of microsporin A. Tetrahedron 2007, 63, 6535–6541.
- Song, K.H.; Oh, S.J.; Kim, S.; Cho, H.; Lee, H.J.; Song, J.S.; Chung, J.Y.; Cho, E.; Lee, J.; Jeon, S.; et al. HSP90A inhibition promotes anti-tumor immunity by reversing multi-modal resistance and stem-like property of immune-refractory tumors. Nat. Commun. 2020, 11, 562.
- Porter, N.J.; Christianson, D.W. Binding of the microbial cyclic tetrapeptide Trapoxin A to the class i histone deacetylase HDAC8. ACS Chem. Biol. 2017, 12, 2281–2286.
- Itazaki, H.; Nagashima, K.; Sugita, K.; Yoshida, H.; Kawamura, Y.; Yasuda, Y.; Matsumoto, K.; Ishii, K.; Uotani, N.; Nakai, H.; et al. Isolation and structural elucidation of new cyclotetrapeptides, trapoxins A and B, having detransformation activities as antitumor agents. J. Antibiot. (Tokyo) 1990, 43, 1524–1532.
- Karpiński, T.; Adamczak, A. Anticancer activity of bacterial proteins and peptides. Pharmaceutics 2018, 10, 54.
- Zhang, H.L.; Hua, H.M.; Pei, Y.H.; Yao, X.S. Three new cytotoxic cyclic acylpeptides from marine Bacillus sp. Chem. Pharm. Bull. (Tokyo) 2004, 52, 1029–1030.
- Kanoh, K.; Matsuo, Y.; Adachi, K.; Imagawa, H.; Nishizawa, M.; Shizuri, Y. Mechercharmycins A and B, cytotoxic substances from marine-derived Thermoactinomyces sp. YM3-251. J. Antibiot. (Tokyo) 2005, 58, 289–292.
- Oberheide, A.; Schwenk, S.; Ronco, C.; Semmrau, L.M.; Gorls, H.; Arndt, H.D. Synthesis, structure, and cytotoxicity of urukthapelstatin A polyazole cyclopeptide analogs. Eur. J. Org. Chem. 2019, 2019, 4320–4326.
- Asolkar, R.N.; Freel, K.C.; Jensen, P.R.; Fenical, W.; Pezzuto, J.M. Arenamides A–C, cytotoxic NFkappaB inhibitors from the marine actinomycete Salinispora arenicola. J. Nat. Prod. 2009, 72, 396–402.