Immunotherapy is a highly emerging form of breast cancer therapy that enables clinicians to target cancers with specific receptor expression profiles. Two popular immunotherapeutic approaches involve chimeric antigen receptor-T cells (CAR-T) and bispecific antibodies (BsAb). Briefly mentioned in this review as well is the mRNA vaccine technology recently popularized by the COVID-19 vaccine. These forms of immunotherapy can highly select for the tumor target of interest to generate specific tumor lysis. Combining emerging immunotherapeutics with tumor marker discovery sets the stage for highly targeted immunotherapy to be the future of cancer treatments. This review highlights the principles of CAR-T and BsAb therapy, improvements in CAR and BsAb engineering, and recently identified human breast cancer markers in the context of in vitro or in vivo CAR-T or BsAb treatment.
- immunotherapy
- breast cancer
- CAR-T
- bispecific antibodies
- mRNA vaccine
- TNBC
- triple-negative breast cancer
- tumor specific/associated antigens
- precision therapy
- immune checkpoints
1. Introduction
Breast cancer is the second leading cause of cancer in women behind skin cancer [1]. Despite trailing behind skin cancer, the lifetime risk of developing breast cancer in females is 1 in 3, which currently leads all cancers [2]. In 2020, breast cancer will lead in new cancer diagnoses among women with an estimated 276,480 women and 2620 men diagnosed with invasive breast cancer [1]. Furthermore, 42,170 women are estimated to die from breast cancer, which is the second highest death total behind lung and bronchus cancer [1][2]. Though 5-year survival rates for stage 1 breast cancer are 85% or above, targeted therapy is needed for aggressive and highly metastatic triple-negative breast cancers that have a median survival of 10 to 13 months [2][3].
Current treatments for breast cancer vary depending on the expression of specific receptors on the surface of the cell. Breast cancer is classified based on expression of receptors which include estrogen receptor (ER), human epidermal growth factor (ERBB2) and progesterone receptor (PR). In general, breast cancer can be broken down into three major categories based on the combination of receptor expression: hormone receptor (ER or PR) positive/ERBB2 negative (HR+/ERBB2–), ERBB2 positive/hormone receptor positive or negative (ERBB2+/HR– or HR+), and triple-negative (HR–/ERBB2–) [3]. The hormone receptors (HR), after binding estrogen or progesterone, function as transcription factors to hundreds of genes implicated in cell division and growth [4]. To classify breast cancer as HR+, greater than 1% of the tumor must stain for either ER or PR [5]. To classify the tumor as ERBB2 positive, ERBB2, also known as HER2, must be overexpressed in the tumor cells [6]. ERBB2 is a 185 kDa transmembrane glycoprotein receptor in the epidermal growth factor (EGFR) family [7].
Activation of ERBB2 receptor triggers intracellular tyrosine kinase activity and a signaling cascade leading to cell survival and metastasis of cancer [8]. In addition to classification by receptor expression, breast cancer can also be classified into ductal or lobular carcinomas [3]. This histological classification depends on whether the cancer’s origin is within the ducts or lobules of the breast. However, classifying breast cancer as HR+/ERRB2–, ERBB2+/HR– or HR+, and triple-negative is vital for targeted therapeutics.
Cancers that are HR+ in premenopausal women are treated with tamoxifen, while HR+ cancers in post-menopausal women are treated with aromatase inhibitors such as anastrozole, exemestane, and letrozole [3]. Tamoxifen is a selective estrogen receptor modulator (SERM) that acts as an estrogen receptor antagonist at the breast, preventing estrogen from binding and performing its normal function [3][9]. Aromatase inhibitors prevent conversion of androgen to estrogen and lower circulating estrogen levels [3][9]. In contrast to HR+ cancers, HR– cancers (ER and PR-) most likely promote cancerous growth and proliferation through alternate pathways. Inhibiting estrogen binding or lowering estrogen production will not be an effective form of treatment in HR– breast cancer. Therefore, treatment may have to target overexpressed ERBB2. Patients with ERBB2+/HR+ cancers are treated with a combination of anti-HR and anti-ERBB2 treatment [3]. Specifically, ERBB2 overexpressing cancers are treated with monoclonal antibodies such as trastuzumab and pertuzumab, which block endogenous ligand binding to ERBB2 receptors [3]. Alternatively, small molecule tyrosine kinase inhibitors such as lapatinib and neratinib can target the intracellular tyrosine kinase domain and inhibit the ERBB2 signaling cascade [3]. Unlike ERBB2+ cancers, those that do not overexpress ERBB2 promote anti-apoptosis and metastasis through other mechanisms and will not respond to ERBB2 inhibitor treatment. ERBB2–/HR– breast cancers are extremely challenging to treat since there are no well-known receptor targets that will slow down the proliferative and metastatic phenotypes of this cancer.
Cancers that are HR–/ERBB2– are referred to as triple-negative breast cancer (TNBC). Since ER, PR, and ERBB2 cannot be targeted for treatment, clinicians will use nonspecific chemotherapeutic agents for treatment [3]. HR+ and ERBB2+ stage 1 breast cancer have a 99% and 95% 5-year survival, respectively, while stage 1 TNBC has an 85% 5-year survival [10]. Metastatic HR+ and ERBB2+ breast cancer has a median overall survival of 4 to 5 years and 5 years, respectively, while metastatic TNBC has a median overall survival of 10 to 13 months [3]. It is evident that TNBC, especially metastatic TNBC, remains highly lethal. Therefore, it is imperative to find new therapies to treat TNBC. Recent studies have found new molecular targets in breast cancer that exhibit selectively lethality to TNBC in vitro and in vivo. This selective lethality arises because the tumor-associated antigen is either lowly expressed or not expressed in normal tissue. Moreover, scientists can harness the power of the immune system via chimeric antigen receptor-T cell therapy (CAR-T therapy) and bispecific antibody therapy (BsAb) to promote immune cell mediated death of cancer cells that highly express the target of interest.
2. Principles of Chimeric Antigen Receptor-T Cell (CAR-T) Therapy
CAR-T cell therapy is designed to target any non-MHC (major histocompatibility complex protein), tumor-bound receptor and is based on the fundamental mechanisms of endogenous T cells. T cells are responsible for recognizing antigens from foreign material or harmful cellular activity, such as pathogens, environmental factors, and tumors (Figure 1) [11]. Antigen-presenting cells (APC), such as macrophages, dendritic cells, and B cells, process foreign invaders and present a piece of their proteins, known as antigen, to inactivated helper T cells via MHC class II [12]. The helper T cell will recognize this antigen with the help of its CD4 co-receptor, which assists in T cell receptor (TCR) binding to the antigen–MHC complex [12]. In addition to T cell receptor binding, the APC displays co-stimulatory proteins that bind to complementary receptors on the T cell to further enable its activation [12]. Upon activation, helper T cells facilitate in the activation of cytotoxic T cells, which are responsible for killing infected cells by binding to antigens presented on MHC Class I protein and inducing apoptosis (Figure 1) [12]. Unlike class II MHC proteins, which are restricted to dendritic cells, macrophages, and B lymphocytes, class I MHC proteins are found on most nucleated cells of the body [12]. T cell activation signaling through the TCR/CD3ζ pathway and through costimulatory receptors, along with much lower affinity for self-peptide-MHC (pMHC), allows for specific CD8+ T cell mediated cytotoxicity (Figure 1) [13].
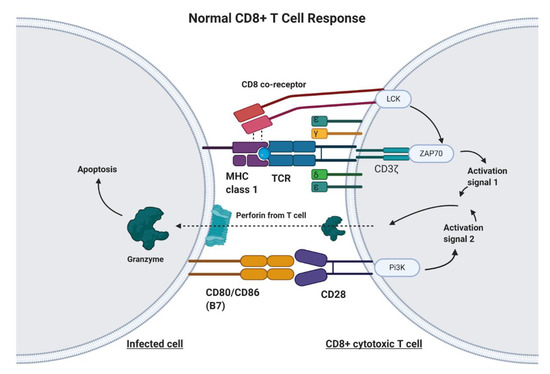
Figure 1. The Normal CD8+ T Cell Response. Infected cells will process and present a portion of the viral or bacterial protein on major histocompatibility complex (MHC) Class 1 molecules. The T cell receptor (TCR) on CD8+ T cells will recognize this antigen bound to MHC. The CD8 co-receptor helps with recognition of MHC Class I. Upon TCR/CD8 co-receptor binding, the intracellular CD8 and CD3ζ domain work together to produce activation signal 1. Additionally, a costimulatory signal involving B7 and CD28 is required in order to promote T cell activation, cytokine production, and prevention of anergy and apoptosis. Upon cytotoxic T cell activation, the T cell releases perforin and granzyme, which act upon the infected cell to initiate the extrinsic apoptosis pathway.
Genetic engineering has utilized the understanding of T cell recognition to create chimeric antigen receptors (CARs) that can be inserted into T cells for specific targeting and lysis [13]. The key component of the CAR is the extracellular single-chain variable fragment (scFv) derived from the fragment antigen binding (Fab) region of immunoglobulin, or another extracellular ligand recognition domain, which defines the T cell’s ability to bind and attack a specific molecular target independent of MHC protein [13]. In addition to the surface domain, CARs are composed of transmembrane and intracellular signaling domains [14]. Contained within these intracellular domains are CD3ζ, CD28, and 4-1BB [15][16]. Normally, T cell receptor heterodimers and CD3ζ join with CD3-gamma, CD3-delta, and CD3-epsilon to form the T cell receptor–CD3 complex [17]. However, the zeta chain specifically plays a role in coupling antigen recognition with several signal transduction pathways, which is the reason for its inclusion in CARs (Figure 1 and Figure 2). Another costimulatory receptor included in the CAR construct, CD28, is critical for T cell activation and operates through CD28 on the T cell interacting with CD80 or CD86 on APCs (Figure 1) [18][19]. CD28 engages the PI3K–AKT pathway to enhance T cell survival, growth, and clonal expansion (Figure 2) [19][20]. The final intracellular domain included in CARs is the 4-1BB costimulatory receptor, which amplifies and diversifies the T cell response after initial activation (Figure 2) [21]. Throughout time, more research and understanding of T cell and costimulatory receptors led to improvements in the CAR construct [22]. In first generation CARs, the scFv (single-chain variable fragment) is joined only by the intracellular CD3ζ domain and proved to be insufficient due to lack of lasting T cell response or sustained cytokine release (Figure 2) [23]. Second generation CAR constructs combined CD3ζ with another costimulatory domain (either CD28 or 4-1BB) to improve activation and survival of the CAR-T cells (Figure 2) [24]. The third generation CAR goes a step further to combine CD3ζ, CD28, and 4-1BB stimulatory domains (Figure 2) [25]. Finally, fourth generation CAR-T cells utilize the third generation CAR construct in addition to transgene expression of costimulatory receptors (CD28, 4-1BB, etc.) or proinflammatory cytokines (Figure 2) [25]. Fourth generation CAR-T cells are currently being heavily investigated and have shown great success in preclinical models [25]. Overall, the CAR is introduced into a patient’s own T cells to allow for maximal T cell activation and specific lysis of tumors via interaction with any tumor-associated or tumor-specific protein independent of MHC presentation [14].
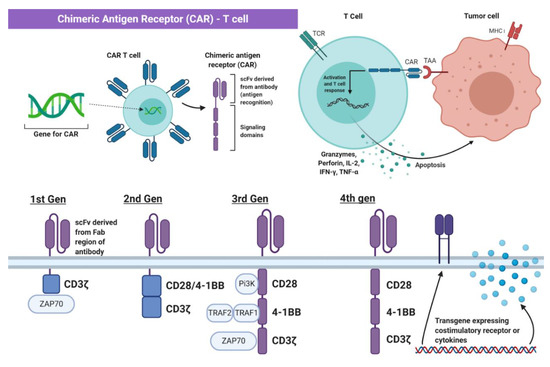
Figure 2. The Chimeric Antigen Receptor T cell (CAR-T Cell). The chimeric antigen receptor (CAR) construct is inserted into T cells and enables initiation of apoptosis via binding to tumor-specific or tumor-associated antigens independent of MHC interaction. Part of the figure adapted from “Chimeric Antigen Receptor (CAR)” and “CAR-Engrafted T cell and Tumor Cell”, by BioRender.com (2021). Retrieved from https://app.biorender.com/biorender-templates (accessed on 28 February 2021).
Creating CAR-T cells from patients’ own leukocytes requires collection, separation, and genetic modification [14]. First, peripheral blood mononuclear cells are extracted from the patient and then the collected apheresis is washed and fractioned [26]. Different devices allow for removing red blood cell and platelet contamination, isolating lymphocytes through size-based fragmentation, and even further isolation of CD4+, CD8+, CD25+, or CD62L+ T cells using beads or chromatography techniques [26]. After the T cells are isolated, they are activated through multiple methods, such as APC or artificial APC based stimulation [26]. Additionally, products such as the Miltenyi MACS GMP TransAct CD3/28 beads, Miltenyi MACS GMP ExpAct Treg beads, Invitrogen CTS Dynabeads CD3/28, and the Juno Stage Expamer technology have been developed to activate and expand the T cells [26].
After collection and separation, the T cells are genetically modified to express the CAR construct through viral transfection, such as σ-retroviral vectors and lentiviral vectors [26]. Retroviral vector transfer utilizes the ability of the virus to convert a single-stranded RNA into a double-stranded DNA that then incorporates itself into the cell genome permanently through transduction [27]. One disadvantage of retroviral vectors is their inability to incorporate into nonreplicating cells [28]. This problem is solved by lentiviral vectors, which are capable of infecting dividing and nondividing cells [29]. Another stable vector comprises the sleeping beauty transposon packaged into a lipid-based vessel [26]. In this system, a transposase enzyme, generated by intracellular machinery transcribing the expression vector then translating to the enzyme, recognizes inverted tandem repeat sequences flanking the transgene on the other vector and then cuts and pastes the gene into TA dinucleotide base pair sites of the T cell genome [30].
Additionally, other methods of generating CAR-T cells have also been developed. Researchers have worked on a protocol involving mRNA electroporation to generate CAR-T cells, which can prevent uncontrolled reactivity caused by viral transductions [31]. Scientists have also used CRISPR to successfully integrate a CD19-specific CAR into the T cell receptor α constant (TRAC) locus, which allowed for robust expression of the CAR and outperformed the function of CARs generated by other methods [32].
In comparison to conventional T cell activity, challenges with safety and specificity arise in CAR-T cells [13]. Unlike conventional T cells, which have a high specificity for their targets, CARs often target both the cells of interest and healthy cells due to the unrestricted nature of certain targets [13]. Researchers have aimed to address this by engineering CAR-T cells that can be eliminated either through conditional suicide gene insertions or through transduced caspase-9, which induces apoptosis upon AP1903 drug administration [13]. It is also possible to target molecules expressed on tumor cells but absent or lowly expressed on healthy cells, such as ROR1 or GD2, or neoantigens present due to oncogenic mutation [13]. An alternative solution could be to transduce T cells with both a CAR and a chimeric costimulatory receptor (CCR) that recognize separate antigens, therefore requiring recognition of two tumor-associated proteins before CAR-T cell activation [33]. Through advanced development of different generations of CAR-T constructs as well as improvements in specificity and activation, CAR-T therapy will undoubtedly be a key component of future targeted immunotherapeutics.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22052433
References
- Breast Cancer Statistics and Resources. Available online: (accessed on 22 December 2020).
- American Cancer Society. Cancer Facts and Figures 2020. Available online: (accessed on 22 December 2020).
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300.
- Brown, M.; Cato, L.; Jeselsohn, R. Chapter 29—Hormone-Responsive Cancers. In Yen and Jaffe’s Reproductive Endocrinology, 8th ed.; Strauss, J.F., Barbieri, R.L., Eds.; Elsevier: Philadelphia, PA, USA, 2019; pp. 717–741. ISBN 978-0-323-47912-7.
- Hammond, M.E.H.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M.; et al. American Society of Clinical Oncology/College of American Pathologists Guideline Recommendations for Immunohistochemical Testing of Estrogen and Progesterone Receptors in Breast Cancer. J. Clin. Oncol. 2010, 28, 2784–2795.
- Wolff, A.C.; Hammond, M.E.H.; Hicks, D.G.; Dowsett, M.; McShane, L.M.; Allison, K.H.; Allred, D.C.; Bartlett, J.M.S.; Bilous, M.; Fitzgibbons, P.; et al. Recommendations for Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Update. J. Clin. Oncol. J. Am. Soc. Clin. Oncol. 2013, 31, 3997–4013.
- Bargmann, C.I.; Hung, M.C.; Weinberg, R.A. The Neu Oncogene Encodes an Epidermal Growth Factor Receptor-Related Protein. Nature 1986, 319, 226–230.
- Yu, D.; Hung, M.-C. Overexpression of ErbB2 in Cancer and ErbB2-Targeting Strategies. Oncogene 2000, 19, 6115–6121.
- Joshi, H.; Press, M.F. 22—Molecular Oncology of Breast Cancer. In The Breast, 5th ed.; Bland, K.I., Copeland, E.M., Klimberg, V.S., Gradishar, W.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 282–307. ISBN 978-0-323-35955-9.
- Chavez-MacGregor, M.; Mittendorf, E.A.; Clarke, C.A.; Lichtensztajn, D.Y.; Hunt, K.K.; Giordano, S.H. Incorporating Tumor Characteristics to the American Joint Committee on Cancer Breast Cancer Staging System. Oncologist 2017, 22, 1292–1300.
- Kumar, B.V.; Connors, T.; Farber, D.L. Human T Cell Development, Localization, and Function throughout Life. Immunity 2018, 48, 202–213.
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002; ISBN 978-0-8153-3218-3.
- Srivastava, S.; Riddell, S.R. Engineering CAR-T Cells: Design Concepts. Trends Immunol. 2015, 36, 494–502.
- Subklewe, M.; Von Bergwelt-Baildon, M.; Humpe, A. Chimeric Antigen Receptor T Cells: A Race to Revolutionize Cancer Therapy. Transfus. Med. Hemother. 2019, 46, 15–24.
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-Lymphocyte Cytotoxicity and Proliferation Directed by a Single Chimeric TCRzeta /CD28 Receptor. Nat. Biotechnol. 2002, 20, 70–75.
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.-H.; Geiger, T.L.; Campana, D. Chimeric Receptors with 4-1BB Signaling Capacity Provoke Potent Cytotoxicity against Acute Lymphoblastic Leukemia. Leukemia 2004, 18, 676–684.
- CD247 Molecule Homo Sapiens (Human) Gene NCBI. Available online: (accessed on 11 January 2021).
- Linsley, P.S.; Ledbetter, J.A. The Role of the CD28 Receptor during T Cell Responses to Antigen. Annu. Rev. Immunol. 1993, 11, 191–212.
- Sayegh, M.H.; Turka, L.A. The Role of T-Cell Costimulatory Activation Pathways in Transplant Rejection. N. Engl. J. Med. 1998, 338, 1813–1821.
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and Regulation of Apoptosis. Biochim. Biophys. Acta BBA Mol. Cell Res. 2011, 1813, 1978–1986.
- Cheuk, A.T.C.; Mufti, G.J.; Guinn, B. Role of 4-1BB:4-1BB Ligand in Cancer Immunotherapy. Cancer Gene Ther. 2004, 11, 215–226.
- Lindner, S.E.; Johnson, S.M.; Brown, C.E.; Wang, L.D. Chimeric Antigen Receptor Signaling: Functional Consequences and Design Implications. Sci. Adv. 2020, 6, eaaz3223.
- Brocker, T.; Karjalainen, K. Signals through T Cell Receptor-Zeta Chain Alone Are Insufficient to Prime Resting T Lymphocytes. J. Exp. Med. 1995, 181, 1653–1659.
- Krause, A.; Guo, H.-F.; Latouche, J.-B.; Tan, C.; Cheung, N.-K.V.; Sadelain, M. Antigen-Dependent CD28 Signaling Selectively Enhances Survival and Proliferation in Genetically Modified Activated Human Primary T Lymphocytes. J. Exp. Med. 1998, 188, 619–626.
- Brentjens, R.J.; Curran, K.J. Novel Cellular Therapies for Leukemia: CAR-Modified T Cells Targeted to the CD19 Antigen. Hematol. Am. Soc. Hematol. Educ. Program 2012, 2012, 143–151.
- Wang, X.; Rivière, I. Clinical Manufacturing of CAR T Cells: Foundation of a Promising Therapy. Mol. Ther. Oncol. 2016, 3, 16015.
- Anson, D.S. The Use of Retroviral Vectors for Gene Therapy—What Are the Risks? A Review of Retroviral Pathogenesis and Its Relevance to Retroviral Vector-Mediated Gene Delivery. Genet. Vaccines Ther. 2004, 2, 9.
- Jolly, D.J. Retroviral Vectors. In Encyclopedia of Cancer, 2nd ed.; Bertino, J.R., Ed.; Academic Press: New York, NY, USA, 2002; pp. 153–166. ISBN 978-0-12-227555-5.
- Patel, D.H.; Misra, A. 5—Gene Delivery Using Viral Vectors. In Challenges in Delivery of Therapeutic Genomics and Proteomics; Misra, A., Ed.; Elsevier: London, UK, 2011; pp. 207–270. ISBN 978-0-12-384964-9.
- Singh, H.; Huls, H.; Kebriaei, P.; Cooper, L.J.N. A New Approach to Gene Therapy Using Sleeping Beauty to Genetically Modify Clinical-Grade T Cells to Target CD19. Immunol. Rev. 2014, 257, 181–190.
- Krug, C.; Wiesinger, M.; Abken, H.; Schuler-Thurner, B.; Schuler, G.; Dörrie, J.; Schaft, N. A GMP-Compliant Protocol to Expand and Transfect Cancer Patient T Cells with MRNA Encoding a Tumor-Specific Chimeric Antigen Receptor. Cancer Immunol. Immunother. CII 2014, 63, 999–1008.
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; Van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC Locus with CRISPR/Cas9 Enhances Tumour Rejection. Nature 2017, 543, 113–117.
- Kloss, C.C.; Condomines, M.; Cartellieri, M.; Bachmann, M.; Sadelain, M. Combinatorial Antigen Recognition with Balanced Signaling Promotes Selective Tumor Eradication by Engineered T Cells. Nat. Biotechnol. 2013, 31, 71–75.