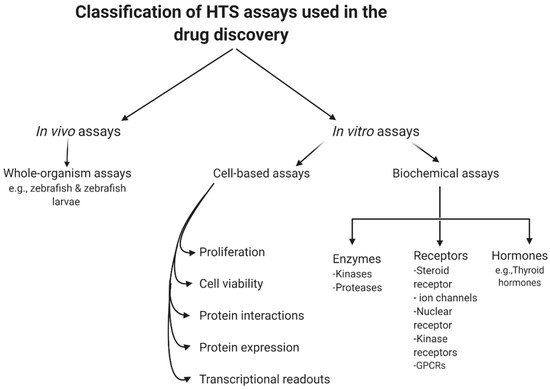
HTS involves in vitro, cell- or whole organism- based assays. The most common readouts for biochemical assays in HTS are optical, including absorbance, fluorescence, luminescence, and scintillation.
1. Formats and Major Considerations for HTS Platforms
HTS involves in vitro, cell- or whole organism- based assays [21]. The most common readouts for biochemical assays in HTS are optical, including absorbance, fluorescence, luminescence, and scintillation. The efficiency of data production and cost per screen are the main determinants in the choice of the most suitable readout for a particular screen. However, the fluorescence-based techniques are considered as one of the primary detection methods [22]. This can mainly be attributed to the high sensitivity and diverse range of available fluorophores enabling multiplexed readouts which allow miniaturization, assay design stability, ease of handling, and the ability to simultaneously track several events in real time [23]. However, it is important to note that short wavelength excitation (particularly those under 400 nm) should be avoided during the development of functional assays in order to reduce interference from test compounds [24,25,26]. This direct screening approach has been applied to the selection of thrombin inhibitors, HIV-protease inhibitors, DNA gyrase inhibitors, etc. [27,28,29]. Quantitative kinetics of compound binding can be used to gain a higher level of understanding of binding mechanisms, as it is possible to investigate the effect of structural variations in a systematic way. Association and dissociation rates can vary independently for a specific lead series, resulting in the rapid evolution of sub nanomolar-affinity leads [30].
Data from screens can be archived and reviewed using information management systems [31] or more laboriously, in Excel spreadsheets. The data is evaluated in order to classify hits: Data points that surpass a certain specified threshold to determine a positive result. Importantly, the threshold limits can be quite subjective, but a value of three standard deviations from the mean signal of wells treated with DMSO, for example, is a fair and typical cut-off, since it offers a manageable false-positive statistical hit rate (0.15%) [32]. Alternatively, the maximum number of hits that can be processed may be increased by “cherry picking”, normally several hundred compounds can be simply picked for further evaluation. Additionally, the median rather than the mean for a single compound can be used to assess hits if the screening is done in triplicate together with the use of appropriate statistical methods [32]. This protects against the undue influence of significant outlier results, which are common in these techniques.
2 Main Types of HTS Assays
2.1. Cell-Based Assays
Using cell-based assays, whole pathways can be investigated generating numerous potential points of interest, as opposed to the analysis of particular predetermined steps as in biochemical assays. Moreover, cell-based assays may provide data that cannot be obtained from a biochemical assay, such as the existence of the pharmacological activity of the screened compound at a particular receptor or the intracellular target [
33,
34]. Consequently, cell-based platforms are especially promising as important tools in the study of cell growth and differentiation, in examining the influence of small molecules and cell growth conditions on cell function and physiology, and also in understanding signaling pathways in mammalian cells. They have also proven to be particularly valuable in studying complex conditions such as CNS injury and NDDs, as many factors can contribute to a specific cellular response [
35].
HTS is frequently accomplished using scaled down cell-based methods. Cell-based tests enable chemical libraries to be tested for molecules that exhibit a diversity of biological activities. In the pharmaceutical industry, cellular microarrays utilizing 96- or 384-well microtiter plates with 2D cell monolayer cultures are commonly used [
35]. Cellular microarrays consist of a solid framework wherein minute volumes of diverse biomolecules and cells can be presented, permitting the multiplexed examination of living cells and, subsequently, the assessment of cellular reactions [
33,
36]. Miscellaneous molecules such as antibodies, polymers and small molecules can be arrayed using automated spotting technology or soft lithography [
37]. Cellular microarrays are also used in small molecule screening [
38,
39]. The screening of small molecules in mammalian cell lines, such as CHO cells, could be considered as an example of utilization of such a system [
40,
41]. There is flexibility in choosing the readout when using a cell-based assay focused on a signaling pathway. For example, if an antibody is available, every stage in which a protein is modified (e.g., phosphorylated), translocated [
42] or changed in its abundance [
43,
44,
45] can be possible readouts [
46]. Multiple NDDs have been studied both with target-based and cell-based screens, including AD [
47], PD [
48], bipolar disease, autism and schizophrenia [
49]. A key feature of cell-based screening is that multiple targets are screened at once, the readout being the outcome of a cellular pathway or network [
50].
2.2. Biochemical Assays
Biochemical screening utilizes a purified target protein of interest and measures the binding of ligands or the inhibition of enzymatic activity in vitro [
51]. These assays are generally conducted in a competition format, in which the compound under study displaces a known ligand or substrate. These assays are typically conducted in 384-well plates, which provide a good compromise between screening volumes (20–50 µL), throughput, and the cost of more sophisticated screening equipment. The readout is typically an optical method such as absorbance, fluorescence or luminescence [
52]. Buratti et al. developed a method in which the activity of a specific RNA binding protein (RBP) (TDP-43) was measured, and due to the established activity of this protein, RBP was shown to be involved in the pathology of PD, AD, and other NDDs [
53]. Additionally, Crowe et al. performed a novel study, screening almost 300,000 compounds to evaluate their effect on tau protein assembly. Formation of toxic tau oligomers in the brain is one of the main observed pathologic events of AD [
54]. Using HTS assays based on complementary thioflavin T fluorescence and fluorescence polarization methods, the effects of inhibitors of tau oligomerization were determined. Specifically, that aminothienopyridazines (ATPZs) caused the inhibition of fibril assembly as well as fibrillization of tau. Additionally, the normal ability of tau to stabilize microtubules was not affected and ATPZs were shown to be promising drugs to treat AD [
54,
55]. Scaling down of bioanalytical activities, in order to decrease production expenses, as well as simplifying transport and saving space in the laboratory has led to a focus on laboratory-on-a-chip technology. Overall, scaling down improves the efficiency of required screening [
56,
57]. However, this could be complicated by extensive time implications, error-recovery rates, and complex experimental design often involving an error-prone robotic operation.
In summary, biochemical assays have the advantage that all hits found are against a known target by design. However, in those situations, the often costly and tedious determination of the molecular mechanisms of action would be needed, even though the target is known. Furthermore, due to the degree to which the predicted target was initially validated in the disease phase, the therapeutic potential of an in vitro hit can still be inconsistent. Even following the determination of such mechanistic details, it is difficult to predict the behavior of such compounds in a more complex cellular environment, due to variability in cellular permeability and metabolism, toxicity, selectivity, and the potential off-target activity of the compound [
58]. However, cell-based assays have the benefit of detecting compounds that affect a phenotype in a complex cellular environment, but still suffer from a poor understanding of the target and mechanism of action. In addition, these experiments are usually more expensive and difficult to conform to miniature HTS assays [
9]. summarizes the current classification of the main HTS assays.
Figure 2. General classification of high-throughput screening (HTS) assays.
3. Economics of HTS
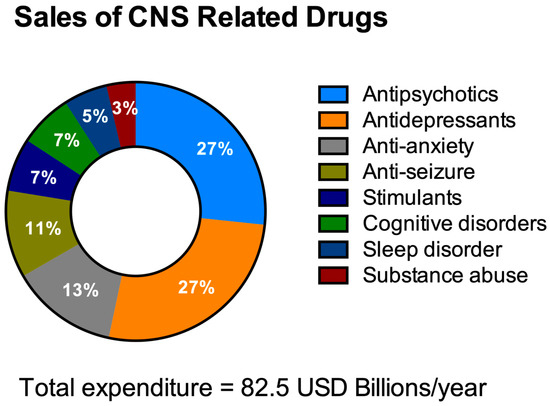
HTS aims to decrease the costs of drug invention [
59,
60]. It is necessary to address the economics of HTS for NDDs drug discovery especially with the escalating yearly costs of mental and neurological pathologies (estimated to be around USD 1 trillion [
61]) including drug sales figures (). It is remarkable to note that 40% of these total costs were attributable to the lack of productivity of the affected population due to the presence of these diseases [
62]. The financial burden of these pathologies is only likely to increase as they typically have long-term consequences combined with an increasingly aging population.
Figure 3. Total expenditure on central nervous system (CNS)-related drugs in 2010. Adapted from Gustavsson et al., 2011 [
77].
It is crucial to extensively enhance our knowledge and understanding of CNS diseases in order to be able to develop effective therapies. Interestingly, despite the number of individuals in the US who experience CNS disorders being more than double than people who suffer from cardiovascular diseases (CVDs), the global market for CNS therapeutics constitutes less than a third of the global drug market for CVDs [
63]. Therefore, the CNS drug market would have to increase by over 5-fold just to correspond to the global market for CVDs.
The primary explanation for this under-development of the worldwide brain drug market is that the vast majority of CNS drugs do not cross the in vivo blood-brain barrier (BBB). The BBB is a unique and highly selective vascular interface that separates the peripheral blood circulation from the neural tissue in order to maintain an optimum homeostatic microenvironment for brain function and protection [
64,
65]. However, biology’s proverbial double-edged sword means that the protective nature of the BBB precludes almost all large-molecule neurotherapeutics and more than 98% of all small-molecules as viable drugs [
66]. In one systematic medicinal research study, over 7000 drugs were evaluated in the comprehensive medicinal chemistry (CMC) database [
67] and this suggested that the CNS was affected by just 5% of these medications. In another study, only one out of every eight medicines analyzed were active in the CNS and only 1% of the total number of drugs were clinically active in the CNS for diseases [
68].
The procedure involved in developing a new drug is an elaborative effort which is often a costly and lengthy process. On average, the cost of developing a new medicine is around USD 1.3 billion (2018) [
69]. However, the expenditure of the research and development (R&D) departments of the major pharmaceutical companies can be as high as USD 2.87 billion (2013) to discover and test a new drug [
70]. Despite these huge investments in new treatments targeting NNDs and an expanding pipeline, there have been more failures and setbacks than overall treatment successes. The failure rate of clinical trials for new treatments targeting NDDs, for example AD, is exceptionally high and usually exceeds 99% [
71]. For example, during the period 2010–2015, all the clinical trials of potential medicines for treating AD failed and were terminated after reaching phase three [
72]. Recently, Biogen terminated both of the phase III, ENGAGE (NCT02477800), and the EMERGE (NCT02484547) clinical trials of Aducanumab [BIIB037], since it failed to demonstrate a superior activity compared to the placebo [
73,
74,
75]. Consequently, Biogen lost more than 5 years and USD 2.5 billion on the failed experimental drug Aducanumab [BIIB037] [
74]. It is clear that R&D expenditures over time have the most impact on the overall cost of drug development [
76].
This entry is adapted from the peer-reviewed paper 10.3390/bioengineering8020030