Sodium glucose co-transporter 2 (SGLT2) inhibitors are effective antihyperglycemic agents by inhibiting glucose reabsorption in the proximal tubule of the kidney.
- SGLT2 inhibitors
1. Introduction
There are six identified SGLT (sodium glucose co-transporter) proteins in humans, of which the SGLT1 and SGLT2 receptors have been studied more thoroughly in recent years. Despite the outstanding sequence similarity between SGLT1 and SGLT2, they show different physiological and biochemical properties. While SGLT1 is primarily expressed in the intestines, SGLT2 is most abundant in the renal cortex, where it plays an essential role in renal glucose reabsorption. SGLT2 inhibitors, including dapagliflozin, canagliflozin, ipragliflozin, tofogliflozin, luseogliflozin, sotagliflozin, ertugliflozin and empagliflozin, have been studied in several clinical studies for the treatment of type 2 diabetes mellitus. Their selectivity for the SGLT2 receptor shows significant variance. While empagliflozin is 2500×, ertugliflozin is 2000×, dapagliflozin is 1200×, canagliflozin is 250×, sotagliflozin is only 20× more selective for the SGLT2 receptor over SGLT1, which may cause significant differences in the mechanism of action [1]. Several studies have proved that SGLT2 inhibitors are associated with reduced cardiovascular morbidity and mortality, including heart failure, and vascular diseases [2][3][4][5][6]. The underlying hypothetic mechanisms of SGLT2 inhibitors beyond their antidiabetic effects are summarized in Figure 1.
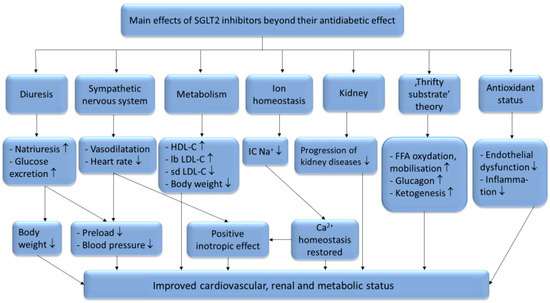
Furthermore, SGLT2 inhibitors have renoprotective effects as well [7]. Besides their beneficial effects on the conventional risk factors for kidney disease (such as blood pressure, hyperglycemia, body weight), it has also been hypothesized that they reduce the intraglomerular pressure [8], change the activation of the renin-aldosterone-angiotensin system [9] and shift renal fuel consumption towards ketone bodies [10].
Interestingly, these cardiovascular and renoprotective effects occur despite an increase in low-density lipoprotein cholesterol (LDL-C) levels which was observed in several clinical studies [11][12]. Nevertheless, this increase in LDL-C happens in the setting of other beneficial changes in plasma lipoprotein metabolism. In this review, we aim to analyze the effects of SGLT2 inhibitors on lipid metabolism to better understand their beneficial effects on cardiovascular outcomes despite slightly elevating LDL-C level.
2. The SGLT2 Inhibitors’ Effect on Plasma Lipoprotein Levels
Several studies reported lowered serum levels of total cholesterol (TC) and triglycerides (TG) [13][14][15] as a result of SGLT2 inhibitor therapy, however there is a debate regarding the changes observed in the serum levels of high-density lipoprotein cholesterol (HDL-C) and low-density lipoprotein cholesterol (LDL-C). According to Calapkulu et al. LDL cholesterol level decreased by 13.4 mg/dL after 6 months in diabetic patients with dapagliflozin (10 mg/die) [13], In contrast, Cha et al. reported an increase of 1.3 mg/dL in LDL level after 24 weeks of dapagliflozin add-on therapy [16], in concordance with the results of Basu et al. in mice [17]. Furthermore, according to Schernthaner et al. canagliflozin (300 mg/die) caused 11.7% increase in LDL after 52 weeks of therapy in patients with type 2 diabetes mellitus [18]. According to Basu et al. the cause of this possible increase in the LDL-C levels could be due to an increased lipoprotein-lipase (LpL) activity and because of a delayed turnover of LDL in the circulation. Canagliflozin reduced the expression of angiopoetin-like protein 4 (ANGPTL4), which is a known inhibitor of LpL in white and brown adipose, skeletal muscle, and heart tissues. With greater LpL activity both the TG and the VLDL levels decreased. They also observed significantly delayed LDL turnover compared to the control group, which can originate from the lowered hepatic levels of the LDL-receptor, which is the major receptor for the clearance of plasma LDL [17]. Another important factor is the ratio of the different LDL subclasses. Using gradient gel electrophoresis (GGE) LDL particles are classified into 4 subclasses, including large (LDL I), intermediate (LDL II), small (LDL III), and very small (LDL IV) LDLs [19][20]. LDL I and II, also referred to as large buoyant (lb) LDL, and LDL III and IV as small dense (sd) LDL particles [21][22]. Small dense LDL particles are more prone to induce metabolic disorders [23][24], obesity [25][26], type 2 diabetes [20][27] and coronary artery disease (CAD) [28] due to their longer circulation time than that of large LDL particles [29], enhanced ability to penetrate the arterial wall and higher susceptibility to oxidation [30]. It is generally known that modified (oxidized and glycated) LDL particles are highly atherogenic and possess more proinflammatory properties than native LDL molecules [28][31]. Interestingly SGLT2 inhibitors slightly increase LDL level yet have beneficial effects on CV morbidity and mortality. The contradiction may be resolved by the results provided by Hayashi et al. showing that dapagliflozin decreased sd LDL, and increased lb LDL levels after 12 weeks of dapagliflozin therapy (5 mg/die) in type 2 diabetic patients [15]. This effect on LDL subclasses ratio may play a significant role in SGLT2 inhibitors’ cardioprotective property [32][33], provided it is a class effect.
Concerning HDL level, according to Kamijo et al. after 12 weeks of canagliflozin administration (100 mg/die) the very large high-density lipoprotein (VLHDL) and large high-density lipoprotein (LHDL) values showed a significant increase, of 10.9% and 11.5% respectively. These beneficial changes might also contribute to subsequent reduction of cardiovascular outcomes, caused by SGLT2 inhibitors [34].
3. The SGLT2 Inhibitors’ Effect on Lipid Regulation
Several signaling molecules have been measured in mice after a 4-week treatment with canagliflozin by Ji et al. [14]. Elevated levels of diacylglycerol O-acyltransferase 2 (DGAT2) mRNA were reversed by canagliflozin. They also observed an increase in peroxisome proliferator-activated receptor-α (PPAR-α), and a decrease in peroxisome proliferator-activated receptor-γ (PPAR-γ) levels [14]. DGAT2 is an integral membrane protein which promotes the synthesis and storage of TG in lipid droplets. The peroxisome proliferator-activated receptors (PPAR) are a group of nuclear receptor proteins that function as transcription factors regulating the expression of several genes. Three types of PPARs have been identified: alpha (α), gamma (γ), and beta/delta (β/δ). While the PPAR-α is expressed mostly in the liver, kidneys, heart, muscle and adipose tissue, it mainly regulates the lipid metabolism in the liver. It is activated under conditions of energy deprivation, and it is necessary for the process of ketogenesis. Activation of PPAR-α promotes the uptake, utilization and catabolism of fatty acids through upregulating the genes that are involved in fatty acid binding and activation, peroxisomal and mitochondrial fatty acid β-oxidation. PPAR-γ regulates the fatty acid storage and glucose metabolism. It activates genes stimulating lipid uptake and adipogenesis in fat cells, it also plays a crucial role in adipocyte differentiation. PPAR-γ increases insulin sensitivity through increasing storage of fatty acids in fat cells, enhancing adiponectin release, inducing fibroblast growth factor 21 (FGF21) and upregulating the cluster of differentiation 36 (CD36) enzyme [35]. This data indicates that canagliflozin suppressed the synthesis of TG and the accumulation of hepatic lipid droplets through the down regulation of DGAT2.
Mice treated with canagliflozin had significant increase in both hepatic and serum FGF21 levels [36]. FGF21 is a fasting-induced hepatokine that stimulates glucose uptake in adipocytes, but not in other cell types. FGF21 acts through the Ras/MAP kinase pathway. This indicates, that canagliflozin triggered a fasting-like catabolic switch, increasing the adipose lipolysis, hepatic fatty acid oxidation and ketogenesis, potentially via FGF21-dependent mechanisms. In addition, FGF21 can induce sympathetic activation in the central nervous system, leading to energy expenditure and weight loss. According to Osataphan et al. FGF21 was essential for the SGLT2 inhibitor induced reduction in adipose tissue mass, adipocyte cell size and activation of lipolysis. While canagliflozin reduced adipocyte size, FGF21-null mice had no weight loss, and adipose tissue and cell size even increased in response to canagliflozin, suggesting that FGF21-null mice had impaired sympathetic and lipolytic activity. FGF21 is also responsible for increasing oxidative metabolism, browning and lipolysis in white adipose tissue [36].
According to the findings of Herrera et al., empagliflozin therapy significantly reduced the gene expression as well as the protein levels of CD36 in atrial tissues of rats after 6 weeks [37]. CD36, also known as fatty acid translocase (FAT), is an integral membrane protein found on the surface of cells that import fatty acids inside cells. It is also a member of the class B scavenger receptor family. CD36 interacts with several ligands, including collagen types I and IV, oxidized LDL and long-chain fatty acids as well. It is also involved in the macrophages’ phagocytosis. After CD36 binds to a ligand, they are internalized thus long-chained fatty acids and oxidized LDL particles can enter the cells. Since autophagy is decreased in metabolic disorders like diabetes and obesity, this dysregulation is an important factor in the pathophysiology of heart failure. The results show, that empagliflozin may ameliorate the impaired basal cardiac levels of autophagy that would lead to the aggregation of proteins, at least in part through CD36, which contributes to the pathogenesis of cardiometabolic diseases.
Xu et al. showed, that empagliflozin elevated AMPK and ACC-CoA phosphorylation in skeletal muscle, and increased hepatic and plasma FGF-21 levels. Empagliflozin also increased energy expenditure, heat production, and the expression of uncoupling protein 1 in brown fat and in inguinal and epididymal white adipose tissue. The M1-polarized macrophage accumulation was reduced, while plasma TNFα levels and obesity-related chronic inflammation decreased. In summary, empagliflozin did not just suppress weight gain by inducing fat utilization and browning, but also attenuated obesity-induced inflammation and insulin resistance [38]. Likewise, Osataphan et al. concluded that canagliflozin therapy activated AMPK through the inhibition of mitochondrial complex I, without an increase in ACC-CoA. However, this change in AMPK phosphorylation was not present in lean mice, thus AMPK is not likely to be the major mediator for the increase in fatty acid oxidation and ketogenesis [36]. One of the key molecules in the transport and oxidation of fatty acids is acetyl-CoA carboxylase (ACC), which converts acetyl-CoA to malonyl-CoA. Malonyl-CoA is an inhibitor of carnitine palmitoyltransferase 1 (CPT-1), which transports fatty acids into the mitochondria for oxidation. Thus, inactivation of ACC results in increased fatty acid transport and subsequent oxidation. On the other hand, AMP-activated protein kinase (AMPK) may decrease malonyl-CoA levels by regulating malonyl-CoA decarboxylase (MCD). Another important role of AMPK is, that it phosphorylates and inactivates 3-hydroxy-3-methylglutaryl-CoA reductase (MHGCR), which is a key enzyme in cholesterol synthesis. AMPK, therefore, regulates fatty acid oxidation and cholesterol synthesis [38]. Mammalian target of rapamycin (mTOR) is a cellular energetic sensor, which is often regulated inversely with AMPK. Osataphan et al. found that after canagliflozin therapy there was a 56% decrease in the hepatic phosphorylation of the mTOR downstream substrate S6 when refeeding in canagliflozin-treated mice. This change was not present in the control groups, thus the decrease in mTOR signaling might be weight-dependent [36].
Empagliflozin therapy reduced cardiac content of sphingolipids (sphingomyelins and ceramides) and glycerophospholipids, which play an important role in connecting insulin resistance to cardiac damage, and even in the development of cardiovascular diseases. It is suggested that changes in lipid metabolism within the heart and cardiac lipid accumulation may have an important role in the development of diabetic cardiomyopathy and heart failure. Ceramides, sphingomyelins and glycerophospholipids are associated with lipotoxicity in the heart, thus they have a major impact on the organ’s functionality. This suggests, that empagliflozin could regulate the metabolism and the cardiac accumulation of these cardiotoxic lipid molecules, which means, that it could be potentially useful for the prevention and treatment of not only diabetic cardiomyopathy, but also for the management of other cardiovascular diseases that have lipotoxicity in their pathogenesis [37].
This entry is adapted from the peer-reviewed paper 10.3390/metabo11020087
References
- Cinti, F.; Moffa, S.; Impronta, F.; Cefalo, M.C.; Sun, A.V.; Sorice, P.G.; Mezza, T.; Giaccari, A. Spotlight on ertugliflozin and its potential in the treatment of type 2 diabetes: Evidence to date. Drug Des. Dev. Ther. 2017.
- Zinman, B.; Inzucchi, E.S.; Lachin, M.J.; Wanner, C.; Ferrari, R.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Kempthorne-Rawson, J.; Newman, J.; et al. Rationale, design, and baseline characteristics of a randomized, placebo-controlled cardiovascular outcome trial of empagliflozin (EMPA-REG OUTCOMETM). Cardiovasc. Diabetol. 2014.
- Guthrie, R. Canagliflozin and cardiovascular and renal events in type 2 diabetes. Postgrad. Med. 2018.
- Wiviott, S.D.; Raz, I.; Bonaca, P.M.; Mosenzon, O.; Kato, T.E.; Cahn, A.; Silverman, G.M.; Zelniker, A.T.; Kuder, F.J.; Murphy, A.S.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019.
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantek, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015.
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017.
- Tsimihodimos, V.; Filippatos, T.D.; Filippas-Ntekouan, S.; Elisaf, M. Renoprotective Effects of SGLT2 Inhibitors: Beyond Glucose Reabsorption Inhibition. Curr. Vasc. Pharmacol. 2017, 15, 96–102.
- Kalra, S.; Singh, V.; Nagrale, D. Sodium-Glucose Cotransporter-2 Inhibition and the Glomerulus: A review. Adv. Ther. 2016.
- Cherney, D.Z.; Perkins, B.A.; Soleymanlou, N.; Maione, M.; Lai, V.; Lee, A.; Fagan, N.M.; Woerle, H.J.; Johensen, O.E.; Broedl, U.C.; et al. Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation 2014, 129, 587–597.
- Mudaliar, S.; Alloju, S.; Henry, R.R. Can a shift in fuel energetics explain the beneficial cardiorenal outcomes in the EMPA-REG OUTCOME study? A unifying hypothesis. Diabetes Care 2016.
- Ptaszynska, A.; Hardy, E.; Johnsson, E.; Parikh, S.; List, J. Effects of dapagliflozin on cardiovascular risk factors. Postgrad. Med. 2013.
- Rodríguez-Gutiérrez, R.; Gonzalez-Saldivar, G. Canagliflozin. Clevel. Clin. J. Med. 2014.
- Calapkulu, M.; Cander, S.; Gul, O.O.; Ersoy, C. Lipid profile in type 2 diabetic patients with new dapagliflozin treatment; actual clinical experience data of six months retrospective lipid profile from single center. Diabetes Metab. Syndr. 2019, 13.
- Ji, W.; Zhao, M.; Wang, M.; Yan, W.; Liu, Y.; Ren, S.; Lu, J.; Wang, B.; Chen, L. Effects of canagliflozin on weight loss in high-fat diet-induced obese mice. PLoS ONE 2017, 12, e0179960.
- Hayashi, T.; Fukui, T.; Nakanishi, N.; Yamamoto, S.; Tomoyasu, M.; Osamura, A.; Ohara, M.; Yamamoto, T.; Ito, Y.; Hirano, T. Dapagliflozin decreases small dense low-density lipoprotein-cholesterol and increases high-density lipoprotein 2-cholesterol in patients with type 2 diabetes: Comparison with sitagliptin. Cardiovasc. Diabetol. 2017.
- Cha, S.A.; Park, Y.M.; Yun, J.S.; Lim, T.S.; Song, K.J.; Yoo, K.D.; Ahn, Y.B.; Ko, S.H. A comparison of effects of DPP-4 inhibitor and SGLT2 inhibitor on lipid profile in patients with type 2 diabetes. Lipids Health Dis. 2017.
- Basu, D.; Huggins, L.A.; Scerbo, D.; Obunike, J.; Mullick, A.E.; Rothenberg, P.L.; Di Prospero, N.A.; Eckel, R.H.; Goldberg, I.J. Mechanism of Increased LDL (Low-Density Lipoprotein) and decreased triglycerides with SGLT2 (sodium-glucose cotransporter 2) inhibition. Arterioscler. Thromb. Vasc. Biol. 2018.
- Schernthaner, G.; Gross, J.L.; Rosenstock, J.; Guarisco, M.; Fu, M.; Yee, J.; Kawaguchi, M.; Canovatchel, W.; Meininger, G. Canagliflozin compared with sitagliptin for patients with type 2 diabetes who do not have adequate glycemic control with metformin plus sulfonylurea: A 52-week randomized trial. Diabetes Care 2013.
- Hirayama, S.; Miida, T. Small dense LDL: An emerging risk factor for cardiovascular disease. Clin. Chim. Acta 2012.
- Berneis, K.; Jeanneret, C.; Muser, J.; Felix, B.; Miserez, A.R. Low-density lipoprotein size and subclasses are markers of clinically apparent and non-apparent atherosclerosis in type 2 diabetes. Metabolism 2005.
- Mikhailidis, D.P.; Elisaf, M.; Rizzo, M.; Berneis, K.; Griffin, B.; Zambon, A.; Athyros, V.; de Graaf, J.; März, W.; Parhoffer, K.G.; et al. “European Panel on Low Density Lipoprotein (LDL) Subclasses”: A Statement on the Pathophysiology, Atherogenicity and Clinical Significance of LDL Subclasses. Curr. Vasc. Pharmacol. 2011, 9, 533–571.
- Berneis, K.K.; Krauss, R.M. Metabolic origins and clinical significance of LDL heterogeneity. J. Lipid Res. 2002, 43, 1363–1379.
- Fan, J.; Liu, Y.; Yin, S.; Chen, N.; Bai, X.; Ke, Q.; Shen, J.; Xia, M. Small dense LDL cholesterol is associated with metabolic syndrome traits independently of obesity and inflammation. Nutr. Metab. 2019, 16, 7.
- Ayyobi, A.F.; McGladdery, S.H.; McNeely, M.J.; Austin, M.A.; Motulsky, A.G.; Brunzell, J.D. Small, dense LDL and elevated apolipoprotein B are the common characteristics for the three major lipid phenotypes of familial combined hyperlipidemia. Arterioscler. Thromb. Vasc. Biol. 2003.
- Magkos, F.; Mohammed, B.S.; Mittendorfer, B. Effect of obesity on the plasma lipoprotein subclass profile in normoglycemic and normolipidemic men and women. Int. J. Obes. 2008.
- Nikolic, D.; Katsiki, N.; Montalto, G.; Isenovic, E.R.; Mikhailidis, D.P.; Rizzo, M. Lipoprotein subfractions in metabolic syndrome and obesity: Clinical significance and therapeutic approaches. Nutrients 2013, 3, 928–948.
- Goldberg, R.; Temprosa, M.; Otvos, J.; Brunzell, J.; Marcovina, S.; Mather, K.; Arakaki, R.; Watsson, K.; Horton, E.; Barrett-Connor, E.; et al. Lifestyle and metformin treatment favorably influence lipoprotein subfraction distribution in the diabetes prevention program. J. Clin. Endocrinol. Metab. 2013.
- Liansheng, W.; Xing, Z.; Yuqi, F.; Fuxiang, C. The detection of serum sdLDL-C in the CAD patients and clinical application. Heart 2011, 97, A244.
- Thongtang, N.; Diffenderfer, M.R.; Ooi, E.M.M.; Barrett, P.H.R.; Turner, S.M.; Le, N.A.; Brown, W.V.; Schaefer, E.J. Metabolism and proteomics of large and small dense LDL in combined hyperlipidemia: Effects of rosuvastatin. J. Lipid Res. 2017, 68, 1315–1324.
- Ohmura, H.; Mokuno, H.; Sawano, M.; Hatsumi, C.; Mitsugi, Y.; Watanabe, Y.; Daida, H.; Yamaguchi, H. Lipid compositional differences of small, dense low-density lipoprotein particle influence its oxidative susceptibility: Possible implication of increased risk of coronary artery disease in subjects with phenotype B. Metabolism 2002.
- Carmena, R.; Duriez, P.; Fruchart, J.C. Atherogenic Lipoprotein Particles in Atherosclerosis. Circulation 2004, 109, III-2–III-7.
- Shiffman, D.; Louie, J.Z.; Caulfield, M.P.; Nilsson, P.M.; Devlin, J.J.; Melander, O. LDL subfractions are associated with incident cardiovascular disease in the Malmö Prevention Project Study. Atherosclerosis 2017.
- Ding, M.; Rexrode, K.M. A Review of Lipidomics of Cardiovascular Disease Highlights the Importance of Isolating Lipoproteins. Metabolites 2020, 10, 163.
- Kamijo, Y.; Ishii, H.; Yamamoto, T.; Kobayashi, K.; Asano, H.; Miake, S.; Kanda, E.; Urata, H.; Yoshida, M. Potential Impact on Lipoprotein Subfractions in Type 2 Diabetes. Clin. Med. Insights Endocrinol. Diabetes 2019, 12.
- Chinetti, G.; Lestavel, S.; Bocher, V.; Remaley, A.T.; Neve, B.; Torrra, I.P.; Teissier, E.; Minnich, A.; Jaye, M.; Duverger, N.; et al. PPAR-α and PPAR-γ activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat. Med. 2001.
- Osataphan, S.; Macchi, C.; Singhal, G.; Chimene-Weiss, J.; Sales, V.; Kozuka, C.; Dreyfuss, J.M.; Pan, H.; Tangcharoenpaisan, Y.; Morningstar, J.; et al. SGLT2 inhibition reprograms systemic metabolism via FGF21-dependent and -independent mechanisms. JCI Insight 2019, 4.
- Aragón-Herrera, A.; Feijói-Bandín, S.; Santiago, M.O.; Barral, L.; Campos-Toimil, M.; Gil-Longo, J.; Pereira, T.M.; García-Caballero, T.; Rodríguez-Segade, S.; Rodríguez, J.; et al. Empagliflozin reduces the levels of CD36 and cardiotoxic lipids while improving autophagy in the hearts of Zucker diabetic fatty rats. Biochem. Pharmacol. 2019.
- Xu, L.; Nagata, N.; Nagashimada, M.; Zhuge, F.; Ni, Y.; Chen, G.; Mayoux, E.; Kaneko, S.; Ota, T. SGLT2 Inhibition by Empagliflozin Promotes Fat Utilization and Browning and Attenuates Inflammation and Insulin Resistance by Polarizing M2 Macrophages in Diet-induced Obese Mice. EBioMedicine 2017.