The multicomponent reaction between phenol, chloroform and acetone is known as Bargellini reaction. In this article we collected and discussed all the Bargellini and Bargellini-like reactions carried out over the years. The main goal is that this knowledge-sharing will help chemists to properly use this transformation and imagine novel reactivities based on it.
- Multicomponent Bargellini Reaction
1. Introduction
1.1. The Discovery of a Novel Reaction
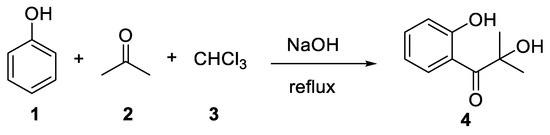
The story of this reaction begins with a German patent published by Link in 1894[1] in which the 2-hydroxy-1-(2-hydroxyphenyl)-2-methylpropan-1-one (4) was prepared by heating a mixture of phenol (1), acetone (2) and chloroform (3) for 5–6 h (Scheme 1).
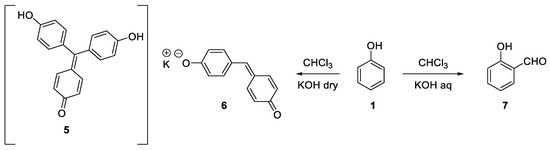
At that time, the reaction between phenols, chloroform and solid sodium hydroxide was already well known. Indeed, it had been reported for the first time in 1872 by Icilio Guareschi, using the pre-formed sodium phenate[2], and later by Sigmund Lustargarden[3]. This reaction was used as a colorimetric method to detect less than 0.1 mg of phenol thanks to the supposed formation of the strongly red-purple colored rosolic acid derivative (5). Actually, the true composition of this precipitate is not known and it could also be the sodium salt of the quinone methide 6 [4][5]. This reaction was later investigated by Reimer and Tiemann who discovered that under aqueous conditions o-formylphenols (7) were obtained (Scheme 2) [6][7].
Although not reported in the original patent, as an archeochemist, it is possible to imagine that acetone was used as an inert solvent by Link, but, contrary to expectations, it reacted. The parallelism with the discovery of the Passerini multicomponent reaction is stunning [8].
However, structure 4 proved to be wrong and 27-year old Guido Bargellini in 1906[9][10] showed that the real product of the reaction was the α-phenoxyisobutyric acid 8 (Scheme 3). Bargellini demonstrated that no phenolic hydroxyl was present as the molecule was neither acetylated nor gave the methyl ether. Moreover, no reactions with hydroxylamine or phenylhydrazine occurred, confirming the absence of the keto group. The compound was soluble in a carbonate solution and it could be re-precipitated by addition of HCl, indicating the presence of a carboxylic acid.

Scheme 3. The reaction revised by Bargellini in 1906.
The same compound 8 had already been obtained in 1900 by Bischoff [11] via basic hydrolysis of the corresponding ethyl ester prepared by reaction between sodium phenate and ethyl 2-bromo-2-methylpropanoate.
From a synthetic point of view, the Bargellini reaction allows for the direct synthesis of aryl ether-bearing sterically hindered carboxylic acids. In the same paper, Bargellini demonstrated the generality of this reaction that works also with other phenolic components, such as α-naphthol, β-naphthol, o- and p-cresol, and thymol. Regarding the mechanism of the reaction, Bargellini proposed the formation of acetonechloroform 9, followed by its hydrolysis to 2-hydroxy-2-methylpropanoic acid 10 and final condensation with phenol 1 (Scheme 4). As we will see later, this hypothesis proved to be wrong, in spite of the fact that it was demonstrated by Bargellini that heating for 6 h in a bain-marie compounds 10 and 1, in the presence of sodium hydroxide, 8 could be obtained in high yield[9].
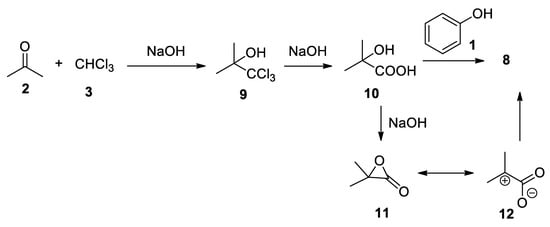
Scheme 4. Sequential reaction mechanism proposed by Bargellini.
In order to explain the reactivity of 2-hydroxy-2-methylpropanoic acid (10), it is possible to consider that 10 under strong basic condition leads to the formation of α-lactone 11. α-Lactones are unstable and reactive intermediates, being attacked by nucleophiles on the α carbon instead of the carbonyl group. This apparently strange behavior can be rationalized considering that α-lactones can be represented by two resonance forms of equal energy, that is, the closed form 11 and an open 1,3-dipolar form 12. High temperatures and polar solvents facilitate the formation of 12 [12][13].
1.2. Guido Bargellini
Guido Bargellini was born in 1879 in Tuscany, more precisely in Roccastrada, a small village near Grosseto, and he took his degree in Chemistry in Rome in 1902. For two years he was laboratory assistant at the University of Siena, and, after a stint in the laboratory of Emil Fisher, he came back to the University of Rome as an assistant, first to Stanislao Cannizaro, and later to Emanuele Paternò. From 1921–1924 he was Professor at the University of Sassari, then of Siena and finally of Rome, where he remained until his death in 1963[14]. His research activity was mainly based on the chemistry of natural substances like santonin, chalcones, flavones, flavanols and phenylcoumarins. At least three eponymous reactions were discovered by Bargellini: (i) the multicomponent reaction described in this review; (ii) a chromatic reaction of polyoxyflavones with sodium amalgam known as Bargellini’s test [15], and (iii) the synthesis of phenylcoumarins by the reaction between sodium phenylacetate and o-hydroxyacetophenones in the presence of acetic anhydride known as Bargellini’s condensation [16][17].
Next to his research activity, he devoted most of his time to the academic teaching, in particular of organic chemistry and war-chemistry (Chimica di Guerra). His book “Lezioni di chimica organica” (Organic chemistry lessons) was one of the first organic chemistry textbooks written by an Italian author. The book had a vast diffusion, reaching the XIth edition and being the leading text for a generation of Italian chemists (Figure 1).

Figure 1. Professor Bargellini and the legendary organic chemistry textbook he authored (this copy belongs to one of the authors (GCT)).
2. The Bargellini Reaction
From now on, we will name Bargellini reactions those transformations in which ketones react with chloroform and then with a nucleophile. Conversely, we will not include in this review those reactions where trichloromethyl carbinols, obtained reacting an aldehyde with chloroform, are used. Indeed, these transformations can be ascribed to other two named reactions: the Jocic-Reeve[18]and the Corey-Link reactions [19]. These transformations have been covered by other recent reviews [20]. In this review, we report, in a chronological order, the most important applications of the Bargellini reaction reported so far, while in the conclusions we provide a practical guide to properly use and essay this multicomponent transformation, together with a perspective on possible unprecedented applications.
Reaction Mechanism and Formation of By-Products
The revised and nowadays accepted mechanism of the Bargellini reaction is due to the work of Weizmann et al. [21]. As already proposed by his inventor, the first step is the formation of 14 which undergoes a fast intramolecular substitution reaction to form the 2,2-dichloro-3,3-dimethyloxirane 15, referred to as the Bargellini epoxide. This epoxide can then undergo a nucleophilic attack by phenolate 16 in an SN2 reaction with substantial SN1 character to form an acyl chloride intermediate 17 which is next hydrolyzed to carboxylate 18 due to the strong basic conditions of the medium (Scheme 5). Seminal contributions by Hine who carried out kinetics experiments on basic hydrolysis of chloroform showed that proton abstraction on chloroform (3) to form the trichloromethane anion 13 is a fast process, while the α-elimination to dichlorocarbene 19 is a slower event which sees the dichlorocarbene in equilibrium with the trichloromethylide anion (Scheme 5)[22][23].
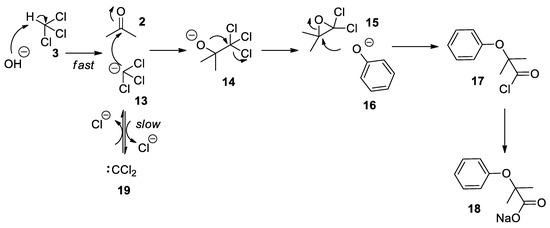
Scheme 5. The most widely accepted mechanism for the Bargellini reaction.
The use of bromoform is possible and it has been reported [24], but chemists should be aware that: (i) dibromocarbene is much more reactive than dichlorocarbene, which alters the equilibrium between these two species, (ii) the basic hydrolysis of CHBr3 is six times faster compared to CHCl3.
The base-induced formation of haloform carbanion follows the order I > Br > Cl > F, while the relative ease to lose a halogen from the trihalocarbanion to give a dihalocarbene is Br > I > Cl > F. Finally, the reactivity of dihalocarbenes follows the following order: F > Cl > Br > I.
After extensive investigations, it is now established that the typical Bargellini conditions require one equiv. of phenol, three equiv. of acetone, four equiv. of chloroform and six equiv. of freshly pulverized sodium hydroxide in THF. Moreover, a careful control of the temperature is mandatory, being the reaction highly exothermic.
Another possible mechanism for the Bargellini reaction, when performed under strict anhydrous conditions, has been proposed and it is based on the reaction of phenolate 16 with acetone 2. The obtained intermediate 20 can react with dichlorocarbene 19, generated in situ by the α-decomposition of the trichloroformate anion, to give 21. An intramolecular rearrangement of 21 gives the corresponding acyl chloride 17 which is immediately hydrolyzed to carboxylate 18 (Scheme 6). To prove the proposed mechanism, the authors reacted phenol with acetone under basic conditions isolating the intermediate 2-phenoxypropan-2-ol [25]. When the latter compound was reacted with chloroform under anhydrous basic conditions, the Bargellini adduct was formed. The authors mention that the presence of water prevents the formation of the product, as dichlorocarbene rapidly reacts with it to form formic acid. Although intriguing, the intramolecular rearrangement of 21 appears unlikely to the authors of this review as the hemiacetal intermediate 2-phenoxypropan-2-ol under basic conditions can re-form acetone and phenolate.
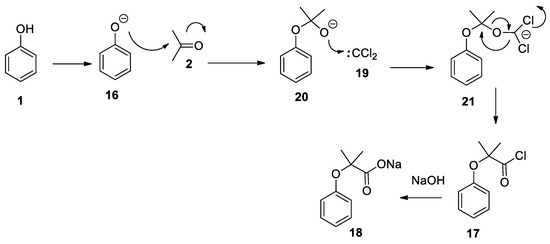
Scheme 6. An alternative, but highly improbable, proposed mechanism of reaction.
It is important to note that the formation of the trichloromethane anion and its attack to acetone to form the oxirane is known to be a very fast reaction, occurring even at low temperature [26], while the formation of dichlorocarbene requires higher temperature.
The Bargellini epoxide 15 can also be formed using the commercially available chlorobutanol 9 (also known as chloretone or acetonechloroform), which has been used in medicine as sedative-hypnotic and bacteriostatic. Treatment of chlorobutanol in the presence of sodium hydroxide generates the gem-dichloroepoxide 15, which can be directly intercepted by nucleophiles. From kinetic data, it has been suggested that Bargellini epoxide might revert to give acetone and dichlorocarbene, which is then hydrolyzed to form carbon monoxide [27][28] (Scheme 7).
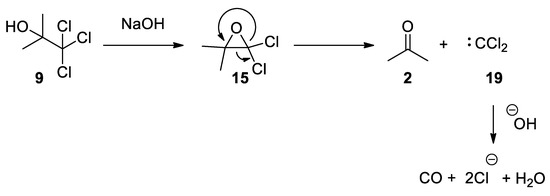
Scheme 7. Commercially available chlorbutanol can be used to form Bargellini epoxide.
Several procedures for the formation of other trichloromethyl carbinols 23 emerged from the literature. For example, aldehydes and ketones can be converted in trichloromethyl carbinols by reacting chloroform, 5% TEBAC and 50% sodium hydroxide. A very fast addition of the base at 0 °C can give higher yields, up to 80% [26], while at reaction temperature highers than 0 °C, the yield is reduced, due to reversion of carbinol 23 to the starting materials 22 and chloroform. Possible by-products deriving from this transformation with ketones are α-hydroxyacids 24, α-chloro acids 25 and α,β-unsaturated acids 26 [29] (Scheme 8).
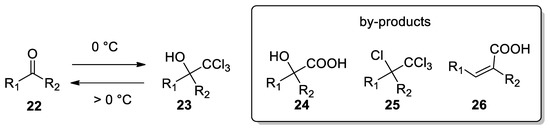
Scheme 8. Careful control of temperature plays a pivotal role in the stability of trichloromethyl carbinol anions and formation of by-products.
In order to avoid the formation of the above-mentioned by-products, 2,2,2-trichloromethyl carbinols 23 have been recently prepared by TBAF-mediated silyl deprotection of trimethyl(trichloromethyl)silane TMSCCl3 previously obtained reacting the ketone with chloroform and LiHMDS. This procedure avoids the use of strong basic conditions [30](Scheme 9).
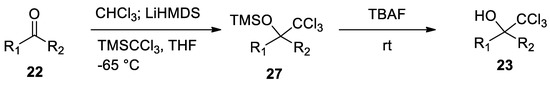
Scheme 9. A novel synthesis of 2,2,2-trichloromethyl carbinols.
Generation of the trichloromethane carbanion under neutral conditions can be also achieved heating at reflux sodium trichloroacetate, which loses carbon dioxide. The carbanion decomposes to dichlorocarbene if there are no acidic protons in the medium[31].
Over the years, other by-products, which can reduce the yield and make the isolation of the desired product difficult, have been isolated from the Bargellini reaction. The awareness of these side-reactions is important in order to minimize their occurrence. A very well-known by-product is for example mesityl oxide 28, which emerges from the self-condensation of acetone. Other reported by-products are those that form from the α-aryloxyisobutyric acid which reacts further with one or two Bargellini epoxides, producing compounds 29 and 30 (Figure 2).
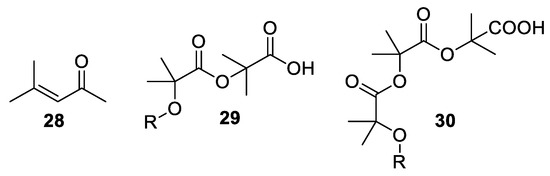
Figure 2. By-products deriving from the Bargellini reaction.
It has been suggested that the nature of the counterion can affect the degree of formation of these by-products. Potassium and tetraalkylammonium carboxylate salts of Bargellini products appear to be more reactive towards Bargellini epoxides, while sodium carboxylate is less prone to react further. LiOH and DBU act similarly to KOH, while t-BuONa leads to an extensive decomposition. In order to avoid the formation of mesityl oxide by-product, the best choice is a slow addition of chloretone solution in acetone to the reaction mixture containing pulverized NaOH, acetone and phenol, under cooling conditions in order to maintain the temperature between 25–30 °C. This represents the best mode of operation in order to maintain high yields and reduce the formation of by-products [31][32].
Recently, dimeric by-products 29 and 30 have been reported as main products with yields ranging from 92 to 41% when refluxing an excess of chloroform, sodium hydroxide and dry acetone for 40–53 h [33].
Use of different carbonyl compounds other than acetone can cause the formation of other specific by-products. For example, with cyclohexanone (31), the Bargellini product 32 was formed together with the 1-chlorocyclohexane-1-carboxylic acid (33). The formation of this by-product is to ascribe to the use of less reactive trapping nucleophiles. (Scheme 10).
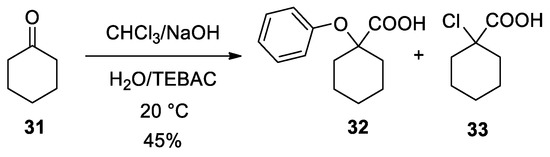
Scheme 10. A by-product (32) which can be obtained from the Bargellini reaction with cyclohexanone when poor nucleophiles are present.
As already discussed above, the trichloromethyl carbinols can decompose if not intercepted by an external nucleophile. Experiments in which different trichloromethyl carbinols reacted in the presence of 10% KOH at 0 °C reveal the formation of the corresponding ketones and carbon monoxide. Again, we note that the source of carbon monoxide is not the trichloromethane anion, but the dichloroepoxide, which slowly breaks down into the carbonyl component and dichlorocarbene that is next hydrolyzed to carbon monoxide [18](Scheme 7 and Scheme 11).
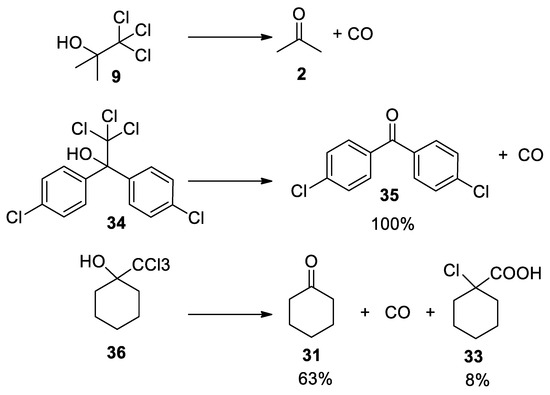
Scheme 11. Decomposition of 2,2,2-trichloromethylcarbinols under basic conditions
3. Applications of Bargellini Reactions
3.1. Reaction with Phenols
After the seminal publication of Bargellini, it was only in 1947 that Galimberti and De Franceschi reported the Bargellini reaction with other phenols [34]. Later, additional works reported the synthesis of dimethylaryloxyacetic acids obtained with other substituted phenols [35][36].
In general, acetone, cyclohexanone, cyclopentanone and methyethylketone are good carbonyl partners in the Bargellini reaction, while benzophenone and acetophenone are not reactive enough.
Clofibric acid is a well-known drug used to reduce the level of triglycerides in the blood. Even if it can be synthesized starting from p-chlorophenol and 2-bromo-2-methylpropanoic acid, [37][38] as an alternative the Bargellini reaction can be used, heating the reagent at reflux for 4 h to yield the desired compound in 37% yield[32][39]. Note that the SN2 reaction between phenols and the sterically demanding 2-bromo-2-methylpropanoic acid is not effective with ortho-substituted phenols, leading to yields ranging from 15 to 60% after prolonged heating under strong basic conditions (even 7 days!). In this case, the Bargellini reaction, which does not suffer from steric hindrance, works much better and should be considered as the first choice [32].
The FT-IT technique was used to monitor the reaction mixture using chlorobutanol and sodium hydroxide. A short-lived species (<5 min) with a unique peak at 758 cm−1 was observed. It has been suggested that this short-lived intermediate is the dimethyl dichloroepoxide[32].
3.2. Reaction with Other Nucleophiles
After the use of phenol and its derivatives, other chemists sporadically reported the successful exploitation of the Bargellini reaction using different types of nucleophiles. Herein, we report the nucleophiles used so far in a chronological order.
In 1929, while studying the Hofmann reaction for the formation of aromatic isocyanides, Gastone Banti from the University of Florence reported the reaction between aniline and chlorobutanol in the presence of sodium hydroxide to afford 2-methyl-2-(phenylamino)propanoic acid. The same conditions were also used for the o-phenyldiamine to provide quinazoline [40]. Unfortunately, this strategy was not studied in detail by the author and this transformation would be re-discovered later, with better reaction conditions and higher yields as described below.
In 1947, besides the reaction with phenols, Galimberti and De Franceschi, in the same publication reported the formation of Bargellini adducts using alcohols (i.e., benzydrol, cyclohexanol), thiols (i.e., p-nitrothiophenol, thio-p-xilenol), 4-methylthiouracil, and benzotriazole [34] (Figure 3). The authors stated that aryl mercaptans reacted better than phenols, but unfortunately no yields were reported in the publication. In another report, thiophenol, p-methoxythiophenol, p-nitrothiophenol and p-methylthiophenol were reacted under Bargellini conditions, yielding the desired α-arylthioisobutyric acids in yields between 72 and 52% [41][42].

Figure 3. Compounds obtained with the Bargellini reaction.
Using acetonechloroform (9), four equiv. of potassium or sodium hydroxide and an aliphatic alcohol 43 at 0 °C, the corresponding Bargellini adducts 44 were obtained in 1948. The reaction worked with primary, secondary and tertiary alcohols, with the primary ones being more reactive. α-alkoxyisobutyric acids can also be dealkoxylated to form metacrylic acids [16] (Scheme 12).
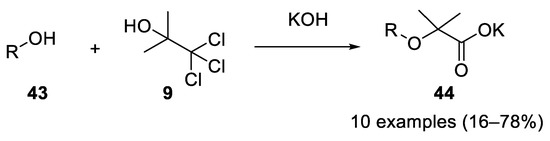
Scheme 12. Aliphatic alcohols as nucleophilic partners in the Bargellini reaction.
Besides acetonechloroform (9), other chlorinated alcohols 45–47 have been used, such as those shown in Figure 4.
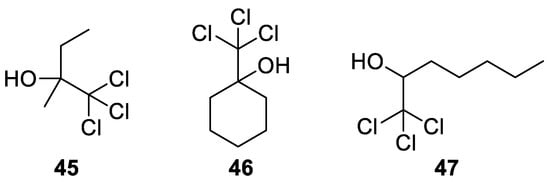
Figure 4. Trichloromethyl carbinols used in the Bargellini reaction with alcohols.
In 1963 thiouracil derivatives 48 and 49 reacted under Bargellini conditions [43] (Scheme 13).
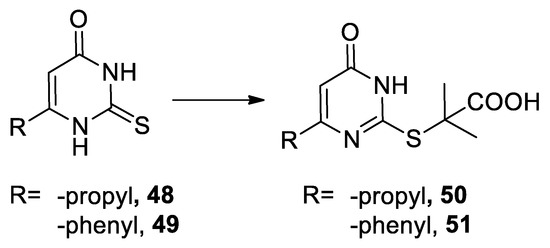
Scheme 13. Reaction with thiouracils.
In 1963 phenyl urea (52) reacted under Bargellini conditions to give two products: an open-chain product 53, which under heating can cyclize affording 3-phenyl-5,5-dimethylhydanthoin 55, and a cyclic product 54 [44](Scheme 14).
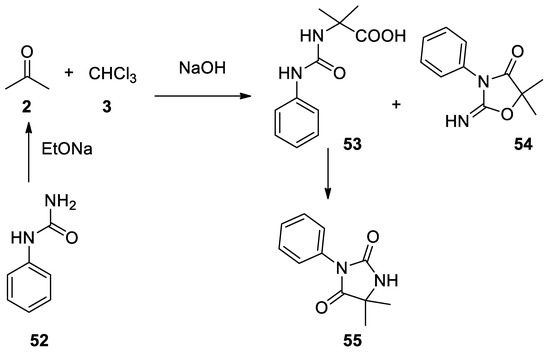
Scheme 14. Reaction with phenylurea.
In the same year the reaction with phenylthiourea (56) was reported. In this case, the two open-chain Bargellini adducts 57–58 were not stable and spontaneously dehydrate to form cyclic compounds 59–60 [45](Scheme 15).
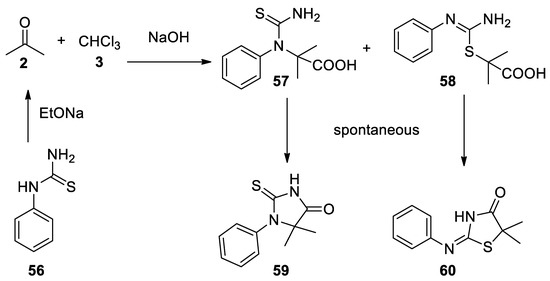
Scheme 15. Reaction with phenylthiourea.
In 1964 semicarbazide (61) and thiosemicarbazides reacted in the Bargellini reaction to give 2-methyl-2-(2-(propan-2-ylidene)hydrazine-1-carboxamido)propanoic acid (62), and the corresponding thio derivative[46](Scheme 16).
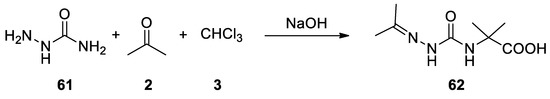
Scheme 16. Reaction with semicarbazide.
A less trivial result was obtained in 1966 reacting guanidine under Bargellini conditions: according to the available spectroscopic methods, the authors stated that compound 63 was obtained. Also, diphenylguanidine and sulfaganidine underwent the Bargellini reaction giving compounds 64 and 65, respectively[47] (Figure 5).
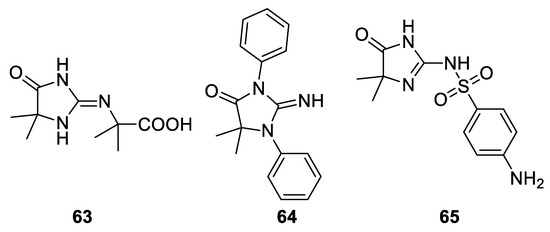
Figure 5. Products obtained from the reaction with guanidine, diphenylguanidine and sulfaguanidine.
In 1980 N-substituted-2-methyl-1,2-propanediamines 66 reacted in the Bargellini reaction in the presence of 50% aqueous sodium hydroxide, chloroform and acetone under phase-transfer catalysis to afford 1,3,3,5,5-pentasubstituted-2-piperazinones 67 [48] (Scheme 17).
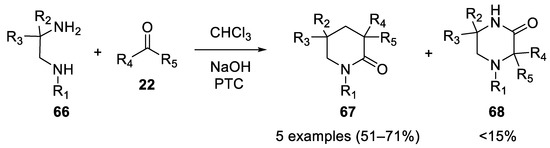
Scheme 17. Reaction with N-substituted-2-methyl-1,2-propanediamines.
The reaction was carried out also using chlorobutanol, giving improved yields. This strategy shows that when a second nucleophilic group is present (as in 69), the ephemeral acyl chloride can be intramolecularly intercepted by the second nucleophile, before being hydrolyzed to carboxylic acid 71. It is possible that the lactam undergoes basic hydrolysis forming the open chain product. On the other hand, it has been shown that electron donating groups on the secondary amine (i.e., 72) are beneficial to the Bargellini reaction, giving a lactam resistant to basic hydrolysis (compound 73, Scheme 18)[49].
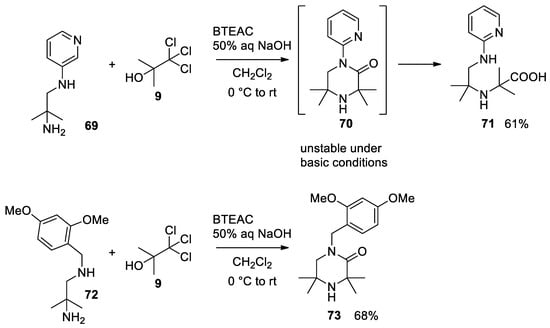
Scheme 18. Bargellini reaction in the presence of a second nucleophilic group.
1,2-Diaminoalchols 74 can also react under classical Bargellini conditions. Dehydration with CSA in toluene forms morpholinone 75. Direct formation of morpholinone, even under phase transfer conditions (PTC), is not possible as the lactone moiety undergoes hydrolysis due to the basic reaction medium that favors the open form 75[50][51][52] (Scheme 19).
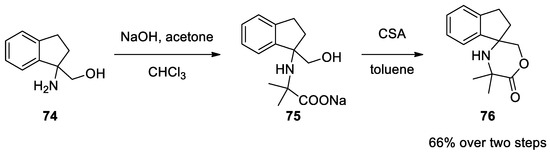
Scheme 19. Reaction with 1,2-diaminoalchols.
In 1980 aliphatic amines 77 were shown to be good partners in Bargellini reactions under PTC conditions. In this case, using an excess of the amine, the intermediate acyl chloride is not hydrolyzed to a carboxylic acid, but rather it reacts with the secondary amines (identical or different from the first one). The net result of this reaction is the formation of hindered acyclic α-aminoacetamides 79. With carbonyl compounds other than acetone, imines are predominantly formed [53] (Scheme 20).
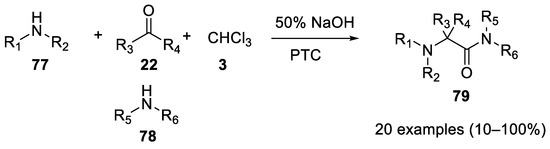
Scheme 20. Reaction with aliphatic amines.
It is interesting to note that under these conditions and for some amines, even acetophenone was reactive. A possible explanation for the formation of the imine when ketones other than acetone are used relies on a different opening mechanism of the Bargellini epoxide. Indeed, when the steric factor is significant, the epoxide can undergo a C-C cleavage, instead of a C-O one, in order to relief the hindrance (Scheme 21).
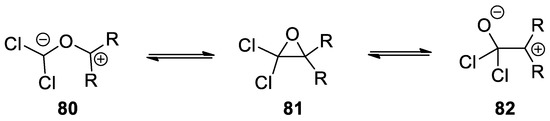
Scheme 21. Resonance forms of the Bargellini epoxide.
Furthermore, it is important to highlight that when aniline 83 and diethylamine 84 were reacted together, contrary to expectations, the less nucleophile aniline attacks first the Bargellini expoxide to form compound 85[53]. To prove this oddity, we carried out this reaction in our laboratory, confirming the result (Scheme 22).
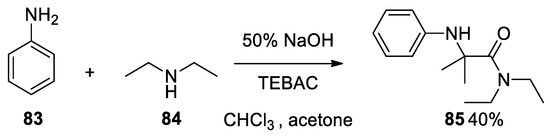
Scheme 22. Bargellini reaction using aniline and diethylamine.
In 1982 o-phenylendiamines 86 were reacted in the Bargellini reaction to form 3,3-dialkyl-1,2,3,4-tetrahydro-2-quinoxalinones 87 under PTC (Scheme 23).
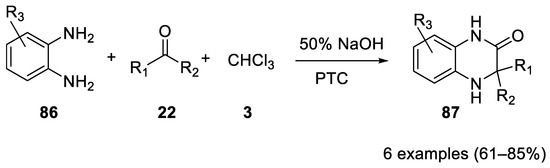
Scheme 23. Reaction with o-phenylendiamines.
Acetone, cyclopentanone, cyclohexanone and octan-2-one react successfully. Similar results were obtained using trichloromethylcarbinol under mild PTC, allowing a high degree of tolerance towards the substituents R1 and R2 [54][55].
In the same paper, the reaction of 1,2-cyclohexyldiamine 88 is reported to form the trans-3,3-dimethyldecahydro-2-quinoxalinones 89 in 75% yield as a mixture of cis and trans isomers 89 (Scheme 24).
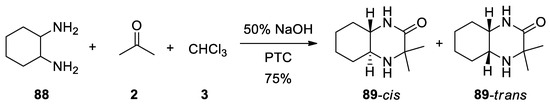
Scheme 24. Reaction with 1,2-cyclohexyldiamine.
In 2001 strongly hindered phenols, such as 2,6-di-tert-butylphenol (90) reacted in the Bargellini reaction with the carbon at the para position, instead of the sterically hindered hydroxy group. The use of PTC conditions allows for the interception of the acyl chloride with an amine group to give 2,6-ditertbutyl-4-(1,1-dialkyl-1-acetamide)-phenols 91. Note that the amines do not compete with the phenol in attacking the oxirane intermediate, allowing for a chemoselective reaction[56](Scheme 25).
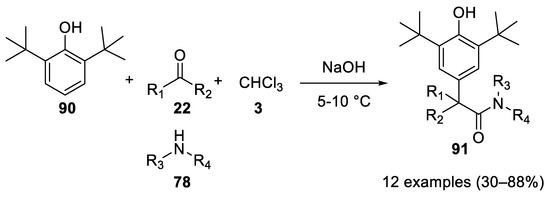
Scheme 25. Reaction with 2,6-ditertbutylphenol.
Although aniline had been shown to react in the Bargellini reaction, no general reaction with other aromatic amines was reported until 2009, despite the seminal contribution of Banti[40]. Different aromatic and heteroaromatic anilines 92 and cyclohexanone or N-Boc-cyclopiperidinone (93) was used and shown to react. The sodium salt precipitates in the reaction medium and the work-up consists only in a simple filtration[57] (Scheme 26). Following this approach, an improved synthesis of carfentanil (96) has been reported (Scheme 26).
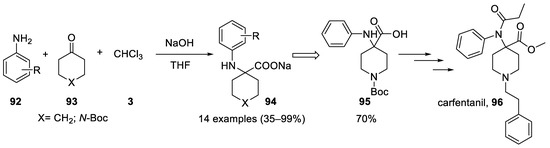
Scheme 26. Reaction with anilines and total synthesis of carfentanil using the Bargellini reaction.
In this paper it was the demonstrated that the poor nucleophilic pyrazole ring 97 reacts under these conditions, affording the Bargellini adduct 98 in good yield (Scheme 27).
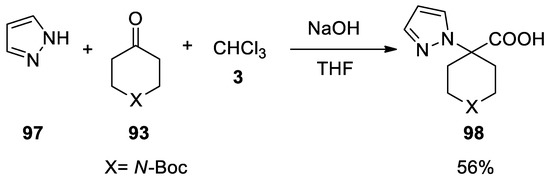
Scheme 27. Reaction with pyrazole.
A method using KF/alumina instead of sodium hydroxide has been reported. Besides anilines, the reaction performed well with phenols and aromatic thiols [58].
As shown in 2012, 1,4-benzodiazepine-3,5-diones (100) can be obtained directly by the Bargellini reaction using 2-amino benzamides 99 as bidentate nucleophile partners [59]. Pre-formation of oxirane followed by the addition of 2-aminobenzamide is mandatory for the success of the reaction. Only acetone and ethylmethylketone were used as carbonyl compounds (Scheme 28).
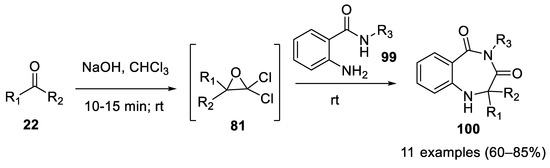
Scheme 28. Synthesis of 1,4-benzodiazepine-3,5-diones using Bargellini reaction.
In 2012 dithiocarbamic acids, prepared in situ from secondary amines 78 and carbon disulfide (101), proved to be valuable nucleophiles in the Bargellini reaction, with acetone, cyclohexanone and cyclopentanone as carbonyl partners [60] (Scheme 29).
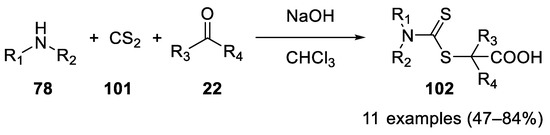
Scheme 29. Reaction with dithiocarbamic acids.
In 2013 ninhydrin (103) proved to be an excellent carbonyl group in the Bargellini reaction with anilines 92 as nucleophiles[61]. The reaction worked with different type of aromatic amines, while with aliphatic ones, such as methyl and ethylamine, yields lower than 20%. Isatin, acenaphthoquinone and 9,10-phenanthrequinone did not react (Scheme 30).
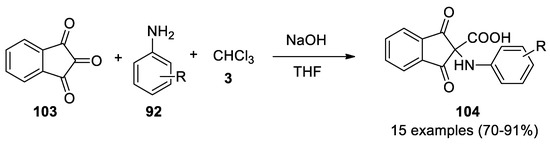
This entry is adapted from the peer-reviewed paper 10.3390/molecules26030558
References
- Link, B. Patent No. 28. R 665-D.R.P 80986; 1894.
- Schiff, H. Correspondenz aus Florenz. Ber. Deut. Chem. Ges. 1872, 5, 1055.
- Lustgarten, S. Über den Nachweis von Jodoform, Naphtol und Chloroform in thierischen Flüssigkeiten und Organen. Mon. für Chem. 1882, 3, 715–722.
- Brieskorn, C.H.; Kallmayer, H.J. Reaktion des Resorcins mit Halogenmethanen unter alkalischen Bedingungen. Arch. Pharm. 1971, 304, 799–804.
- Brieskorn, C.H.; Kallmayer, H.J. Reaktionen der Naphthole mit Halogenmethanen unter alkalischen Bedingungen. Arch. Pharm. 1972, 305, 141–148.
- Reimer, K.; Ferd, T. Ueber die Einwirkung von Chloroform auf alkalische Phenolate. Chem. Ber. 1876, 9, 824–828.
- Wynberg, H. The Reimer-Tiemann Reaction. Chem. Rev. 1960, 60, 169–184.
- Giustiniano, M.; Basso, A.; Mercalli, V.; Massarotti, A.; Novellino, E.; Tron, G.C.; Zhu, J. To Each his Own: Isonitriles for All Flavors. Functionalized Isocyanides as Valuable Tools in Organic Synthesis. Chem. Soc. Rev. 2017, 46, 1295–1357.
- Bargellini, G. Azione del Cloroformio e Idrato Sodico sui Fenoli in Soluzione nell’cetone. Gazz. Chim. Ita. 1906, 36, 329–338.
- Papeo, G.; Pulici, Italian Chemists’ Contributions to Named Reactions in Organic Synthesis: An Historical Perspective. Mol-ecules 2013, 18, 10870–10900.
- Bischoff, C.A. Studien über Verkettungen. XLIII. α-Phenoxy-Buttersäure, -Isobuttersäure und -Isovaleriansäure und deren Ester. Ber. Deut. Chem. Ges. 1900, 33, 931–939.9.
- Wheland, R.; Bartlett, P.D. Alpha-Lactones from Diphenylketene and Di-tert-butylketene. J. Am. Chem. Soc. 1970, 92, 6057–6058.
- Ruggiero, G.D.; Williams, I.H. Oxiranones: α-lactones or zwitterions? Insights from calculated electron density distribution analysis. J. Chem Soc. Perkin Trans. 2 2001, 733–737, doi:10.1039/B100587
- Marini-Bettolo, G.B. Necrologio. Chim. e l’Industria 1963, XLV, 1558–1559.
- Bargellini, G. 1,2,3-Trihydroxyflavone. Contribution to the knowledge of the constitution of scutellarein. Gazz. Chim. Ita. 1919, 49, 47–63.
- Bargellini, G. Fenilcumarine. Gazz. Chim. Ita. 1925, 55, 945–951.
- Wang, Z. Bargellini Condensation. In Comprehensive Organic Name Reactions and Reagents; Wang, Z., Ed.; Wiley: Hoboken, NJ, USA, 2010.
- Reeve, W.; McKee, J.R.; Brown, R.; Lakshmanan, S.; McKee, G.A. Studies on the Rearrangement of (Trichlorome-thyl)carbinols to α-Chloroacetic Acids. Can. J. Chem. 1980, 58, 485–493.
- Corey, E.J.; Link, J.O. A General, Catalytic, and Enantioselective Synthesis of α-Amino Acids. J. Am. Chem. Soc. 1192, 114, 1906–1908.
- Snowden, T.S. Recent Applications of Gem-Dichloroepoxide Intermediates in Synthesis. Arkivoc 2012, 2, 24–40.
- Weizmann, CH.; Sulzbacher, M.; Bergmann, E. The Synthesis of α-Alkoxyisobutyric Acids and Alkyl Methacrylates from Acetonechloroform. J. Am. Chem. Soc. 1948, 70, 1153–1158.
- Hine, J. Carbon Dichloride as an Intermediate in the Basic Hydrolysis of Chloroform. A Mechanism for Substitution Reac-tions at a Saturated Carbon Atom. J. Am. Chem. Soc. 1950, 72, 2438–2445.
- Moss, R.A. “Carbon Dichloride”: Dihalocarbenes Sixty Years After Hine. J. Org. Chem. 2010, 75, 5773–5783.
- Porcelli, L.; Gilardi, F.; Laghezza, A.; Piemontese, L.; Mitro, N.; Azzariti, A.; Altieri, F.; Cervoni, L.; Fracchiolla, G.; Giudici, M.; et al. Synthesis, Characterization and Biological Evaluation of Ureidofibrate-Like Derivatives Endowed with Peroxi-some Proliferator-Activated Receptor Activity. J. Med. Chem. 2012, 55, 37–54.
- Rehman, M.Z.; Kashmiri, M.A.; Ahmed, V.U.; Kazi, A.A.; Siddiqui, H.L. An Efficient Method for the Synthesis of Alkyl 2-(4-benzoylphenoxy)-2-methyl Propanoates. J. Chem. Soc. Pak. 2007, 29, 352–356.
- Merz, A.; Tomahogh, R. Zur Reaktion des Makosza-Reagens mit Aldehyden und Ketonen. Chem. Ber. 1977, 110, 96.
- Nair, A.D.; Lach, J.L. The Kinetics of Degradation of Chlorobutanol. J. Am. Pharm. Assoc. 1959, 48, 390–395.
- Bressanin, G.; Segre, G. Action of Alkaline Solutions on Trichloro Organic Compounds. Gazz. Chim. Ita. 1911, 41, 671–674.
- Kuhl, P.; Muhlstadt, M.; Graefe, J. Phasentransfer-katalysierte Reaktionen; Vl1. Synthese von 1-Chlorocyclohexancarbonsäure aus Cyclohexanon. Synthesis 1976, 12, 825–826.
- Henegar, K.E.; Lira, R. One-Pot in Situ Formation and Reaction of Trimethyl(trichloromethyl)silane: Application to the Synthesis of 2,2,2-Trichloromethylcarbinols. J. Org. Chem. 2012, 77, 2999–3004.
- Verhoek, F.H. The Kinetics of the Decomposition of the Trichloroacetates in Various Solvents. J. Am. Chem. Soc. 1934, 56, 571–577.
- Cvetovich, R.J.; Chung, J.Y.L.; Kress, M.H.; Amato, J.S.; Matty, L.; Weingarten, M.D.; Tsay, F.-R.; Li, Z.; Zhou, G. An Efficient Synthesis of a Dual PPAR α/γ Agonist and the Formation of a Sterically Congested α-Aryloxyisobutyric Acid via a Bargellini Reaction. J. Org. Chem. 2005, 70, 8560–8563.
- Nagan Nirmalan, K.; Ramalakshmi, N. Synthesis of Novel Phenoxy—Isobutyric Acid Derivatives, Reaction of Ketone under Bargellinic Reaction Conditions. Int. J. Chem. Tech. Res. 2015, 8, 1248–1252.
- Galimberti, P.; De Franceschi, A. Sintesi di Alcuni Derivati α-Isobutirrici. Gazz. Chim. Ita. 1947, 77, 431–438.
- Gilman, H.; Wilder, G.R. Some Substituted α-(Aryloxy)-isobutyric Acids and Amides. J. Am. Chem. Soc. 1955, 77, 6644–6646.
- Julia, M.; Baillarge, M.; Tchernoff, G. Sur Quelques nouveaux dérivés aryloxyisobutyriques et apparentés. Bull Soc Chim France 1956, 776–783..
- Davis, R.D.; Fitzgerald, R.N.; Guo, J. Improved Method for the Synthesis of 2-Methyl-2-Aryloxypropanoic Acid Derivatives. Synthesis 2004, 12, 1959–1962.
- Anderson, J.E.; Davis, R.; Fitzgerald, R.N.; Haberman, J.M. Selective Phenol Alkylation: An Improved Preparation of Efaproxiral. Synthetic Commun. 2006, 36, 2129–2133.
- Bose, A.K.; Maghar, S.M.; Ganguly, S.N.; Pednekar, S.; Mandadi, A. Water-Based Biphasic Media for Exothermic Reactions: Green Chemistry Strategy for the Large Scale Preparation of Clofibric Acid and Analogues. Tetrahedron Lett. 2005, 46, 3011–3013.
- Banti, G. Azione del Cloretone e Potassa sulle Basi Aromatiche Primarie. Gazz. Chim. It. 1929, 59, 819–824.
- Andreani, F.; Andrisano, R.; Andreani, A. New α-substituted aryl thioacetic derivatives forming analogues of clofibrate Il Farmaco 1975, 30, 847–858.
- Melnikova, Y.V.; Lyakhov, A.S.; Ivashkevich, L.S.; Artamonova, T.V.; Novoselov, N.P.; Zevatskii, Y.E.; Myznikov, L.V. The Bargellini reaction in a series of Heterocyclic Thiols. Russ. J. Gen. Chem. 2016, 86, 312–316.
- Carminati, G.; Galimberti, P.; Melandri, M. Su Alcuni Derivati α-Isobutirrici. Boll. Chim. Farm. 1963, 102, 522–540.
- Buttini, A.; Galimberti, P.; Gerosa, V.; Melandri, M. Nuove Sintesi di Derivati Idantoinici. Boll. Chim. Farm. 1963, 102, 91–98.
- Buttini, A.; Galimberti, P.; Gerosa, V. Nuova Sintesi di Derivati Tiazolidonici. Boll. Chim. Farm. 1963, 102, 156–160.
- Buttini, A.; Melandri, M.; Galimberti, P. Introduzione del Radicale alfa-Isobutirrico su Semicarbazide, Tiosemicarbazide e Relativi 1 e 4 Fenilderivati. Ann. Chim. 1964, 54, 1112–1121.
- Melandri, M.; Buttini, G.; Gallo, G.; Pasqualucci, C.R. New Synthesis of the Glycocyamidine Group. Ann. Chim. 1966, 56, 1259–1266.
- Lai, J.T. Hindered Amines. Novel Synthesis of 1,3,3,5,5-Pentasubstitued 2-Piperazinones. J. Org. Chem. 1980, 45, 754–755.
- Alanine, T.A.; Stokes, S.; Scott J.S. Synthesis of Electron-Deficient N1-(Hetero)aryl 3,3,5,5-Tetramethyl Piperazinones. Synlett 2017, 28, 357–361.
- Rychnovsky, S.D.; Beauchamp, T.; Vaidyanathan, R.; Kwan, T. Synthesis of Chiral Nitroxides and an Unusual Racemization Reaction. J. Org. Chem. 1998, 63, 6363–6374.
- Nesvadba, P. Radicals and Polymers. Chimia 2018, 72, 456–476.
- Nesvadba, P.; Kramer, A.; Zink, M.-O. Heterocyclic alkoxyamine polymerization composition useful as regulator in free-radical polymerization to obtain low-polydispersity polymeric resins. DE19949352A1, 2000.
- Lai, J. T. Hindered Amines. 2. Synthesis of Hindered Acyclic α-Aminoacetamides. J. Org. Chem. 1980, 45, 3671–3673.
- Alanine, T.A.; Stokes, S.; Scott, J.S. Practical Synthesis of 3,3-Substituted Dihydroquinoxalin-2-ones from Aryl 1,2-Diamines using the Bargellini Reaction. Tetrahedron Lett. 2016, 57, 4386–4388.
- Kristoffersen, T.; Hansen, J.H. 3,4-Dihydroquinoxalin-2-ones: Recent Advances in Synthesis and Bioactivities (microreview). Chem. Heterocycl. Com. 2017, 53, 310–312.
- Lai, J.T. Totally Hindered Phenols. 2,6-Di-t-butyl-4-(1,1-dialkyl-1-acetamide)-phenols and their Persistent Phenoxy Radicals. Tetrahedron Lett. 2001, 42, 557–560.
- Butcher, K.J.; Hurst, J. Aromatic Amines as Nucleophiles in the Bargellini Reaction. Tetrahedron Lett. 2009, 50, 2497–2500.
- Rohman, M.d.R.; Myrboh, B. KF-Alumina-Mediated Bargellini Reaction. Tetrahedron Lett. 2010, 51, 4772–4775.
- Mahdavi, M.; Asadi, M.; Saeedi, M.; Rezaei, Z.; Moghbel, H.; Foroumadi, A.; Shafiee, A. Synthesis of Novel 1,4-Benzodiazepine-3,5-dione Derivatives: Reaction of 2-Aminobenzamides under Bargellini Reaction Conditions. Synlett 2012, 23, 2521–2525.
- Aryanasab, F.; Saidi, M.R. Dithiocarbamic Acids and Thiols as Nucleophiles in the Bargellini Reaction. Sci. Iran. 2012, 19, 551–554.
- Rashid, Z.; Ghahremanzadeh, R.; Naeimi, H. Bargellini Condensation of Ninhydrin as a Ketone and Substituted Anilines as Nucleophiles. New J. Chem. 2016, 40, 1962–1965.