Metabolism is considered to be the core of all cellular activity. Thus, extensive studies of metabolic processes are ongoing in various fields of biology, including cancer research. Cancer cells are known to adapt their metabolism to sustain high proliferation rates and survive in unfavorable environments with low oxygen and nutrient concentrations. Hence, targeting cancer cell metabolism is a promising therapeutic strategy in cancer research. However, cancers consist not only of genetically altered tumor cells but are interwoven with endothelial cells, immune cells and fibroblasts, which together with the extracellular matrix (ECM) constitute the tumor microenvironment (TME). Cancer-associated fibroblasts (CAFs), which are linked to poor prognosis in different cancer types, are one important component of the TME. CAFs play a significant role in reprogramming the metabolic landscape of tumor cells, but how, and in what manner, this interaction takes place remains rather unclear.
- cancer
- cancer-associated fibroblasts (CAFs)
- CAF-tumor metabolic cross-talk
- tumor metabolism
- metabolomics’ measurement techniques
- in silico modeling
- personalized metabolic drugs
1. Introduction
1.1. CAFs as the Epitome of Tumor Metabolism
Max Borst first noted the importance of the tumor microenvironment (TME) on cancer progression in 1902 [1]. Today, it is well acknowledged that the tumor mass includes not only a highly heterogenous cancer cell population but also various types of resident and infiltrating host cells. Cancer-associated fibroblasts (CAFs) are a major component of the TME. CAFs are fibroblasts that display an activated phenotype: they tend to be larger than their normal counterparts, are spindle-shaped and show the presence of stress fibers. This phenotype is transiently observed in normal fibroblasts during wound healing. In contrast, CAFs seem to be constantly activated and unable to revert to a quiescent phenotype [2]. This observation contributed to Harold Dvorak’s definition of cancer as “the wound that does not heal” [3]. Additionally, the CAF genome was reported to be subject to epigenetic reprogramming whereas most studies have not found mutations in CAFs [4].
CAFs communicate with cancer cells in various ways in supporting tumorigenesis. These include signaling molecules, secretion of growth factors, interleukins and metabolite exchanges [5]. CAFs secrete extracellular matrix components such as collagen and laminins, and produce a plethora of cytokines and chemokines (e.g., interferon-γ, stromal cell-derived factor-1 (SDF-1) commonly known as C-X-C Motif Chemokine Ligand 12 (CXCL12), TNF-α and several interleukins), as well as growth factors (e.g., nodal, transforming growth factor β (TGFβ), fibroblast growth factor (FGF)) [6][7][8]. CAFs also secrete pro-angiogenic factors like vascular endothelial factor (VEGF) and endothelial growth factor (EGF). Most of these factors display pro-tumorigenic effects leading to disease progression. Consequently, the past two decades witnessed the development of novel approaches, which focused on targeting stromal cells. Significant reviews have extensively described the stroma–CAFs–cancer cell interactions [9][10][11][12][13][14][15][16]. Nevertheless, metabolic reprogramming by these tumor–stroma interactions was barely addressed so far and rarely considered as a novel therapeutic avenue.
Thus, we focus in this review on our current understanding of the mechanisms underlying cancer cell–CAF metabolic interactions, which need further investigation in order to develop novel therapies. We discuss in detail, the activation of different metabolic pathways, which results from the interaction between tumor cells and CAFs. Distinct metabolic analyses on the tumor-CAF crosstalk, covering in vitro, in vivo and in silico modelling approaches, are described and critically evaluated. We present the idea of mapping the complex metabolic landscape of tumor cells and CAFs in order to reach mechanistic insights, by the integration of multi-omics data into context-specific metabolic models. Finally, we discuss the future development of therapeutic strategies considering metabolic targets identified in CAFs.
1.2. CAF Heterogeneity: The Different CAF Subtypes
There are several theories regarding the origins of CAFs (Figure 1). A first theory suggests that CAFs may derive from the reprograming (and also metabolic rewiring) of normal resident fibroblasts, triggered by bone marrow-derived mesenchymal stem cells [17] or from cancer cells [18][19]. A second theory proposes that CAFs may derive, at least partially, from cancer-associated adipocytes or their progenitor stem cells when exposed to cancer cells [20]. Third, CAFs could originate from both mesenchymal and hematopoietic stem cells, however, the process of this differentiation is still under study [21]. A fourth possibility would be that CAFs originate from epithelial cells through a process called “epithelial to mesenchymal transition” or, similarly, from endothelial cells through “endothelial to mesenchymal transition” [22]].
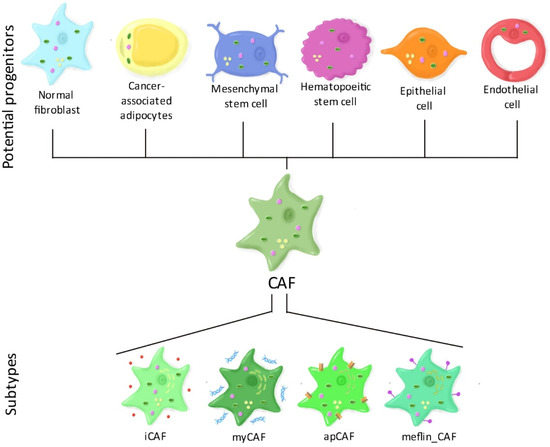
Figure 1. CAF heterogeneity. The origin of cancer-associated fibroblasts (CAFs) remains unclear. Several potential progenitors have been identified including normal fibroblasts, cancer-associated adipocytes, mesenchymal or hematopoietic stem cells, epithelial cells, and endothelial cells. Such heterogeneity in progenitors suggest that CAFs consist of several populations. Today, two principal CAF subtypes have been established: Inflammatory fibroblasts (iCAF) that secrete high levels of cytokines, and myofibroblasts (myCAF) that secrete extracellular matrix components. Additionally, new putative subtypes have been suggested such as antigen-presenting CAFs (apCAF) that express major histocompatibility complex (MHC) class II genes and could be responsible for CD4+ T-cells deactivation, and meflin-expressing CAFs (meflin-CAF), which were found to reduce tumor progression.
Such heterogeneity in their origin suggests that CAFs consist of several subpopulations. Alternatively, it is possible that CAFs respond to specific signals at different topological sites within the TME, leading to different activation states of CAFs and thus contributing to CAF heterogeneity. Today, four putative CAF subtypes have been identified, especially in pancreatic cancer. Myofibroblasts (myCAFs) express high levels of α-SMA and contractile proteins. They are mostly involved in extracellular matrix remodeling, muscle contraction and focal adhesion. Inflammatory fibroblasts (iCAFs) secrete high amounts of cytokines such as IL-6 and IL-8. They also strongly synthesize matrix proteins, especially hyaluronan and are involved in various inflammatory pathways such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and janus kinase/signal transducers and activators of transcription (JAK/STAT) signaling [23]. Interestingly, myCAFs and iCAFs have different effects on the response to immunotherapy. myCAFs were found to be responsible for primary immunosuppression by increasing the infiltration of regulatory T-cells and reducing effector T-cell infiltration, whereas iCAFs were associated with an immunocompetent environment in breast cancer [24]. Recently, new putative subtypes have been suggested. Antigen-presenting CAFs (apCAFs) express major histocompatibility complex (MHC) class II genes and could be responsible for CD4+ T-cells deactivation [25]. Finally, CAFs that express meflin (meflin_CAFs), a membrane-anchored protein were found to decrease tumor progression in pancreatic cancer [26].
Recently, Kieffer and colleagues suggested that in breast and ovarian cancer, CAFs can also be separated into four different subsets according to their location and the expression of specific markers including FAP, α-SMA and CD29 [24]. While two of these subsets (2 and 3) are also found in healthy tissues, subset 1 and 4 are specific to tumors and lymph nodes and are involved in the metastatic process. The subset 1 is additionally involved in immunotherapy resistance [24][27][28].
However, the impact of CAF heterogeneity on tumor progression remains widely unknown. It is tempting to speculate that metabolic modelling coupled with single cell RNA (scRNA) sequencing will allow shedding light on the metabolic differences between the CAF subtypes.
2. Tumor Cell/CAF Interaction-Driven Metabolic Rewiring in Cancer
CAFs have been considered to act as major regulators in shaping tumor metabolism especially through the dysregulation of several metabolic pathways including glucose, amino acid and lipid metabolism [27][28]. The orchestration of these metabolic switches is believed to shape distinct CAF behavior and change tumor cell behavior by these CAFs.
2.1. Glucose Metabolism and Other Sugar Metabolism
In the presence of O2, cells utilize glycolysis to catabolize glucose into pyruvate. The generated pyruvate enters the tricarboxylic acid (TCA) cycle, where it is further oxidized and used to produce energy through oxidative phosphorylation (OXPHOS). In anaerobic conditions, cells oxidize glucose by converting it to lactate, due to the lack of O2, they are unable to utilize OXPHOS. This is a very inefficient process and cells normally only convert a very small amount of glucose to lactate. However, in a phenomenon first identified in the early 20th century by Otto Warburg, cancer cells in culture appear to be reducing a significant percentage of glucose to lactate even in the presence of O2. This phenomenon, known as the “Warburg Effect” [29], has been extensively studied and believed to hold true in in vitro models [30][31]. In line, some cancer types in vivo, such as renal cell carcinomas, clearly rely on aerobic glycolysis to generate energy [32]. However, recent findings have demonstrated that this is not a common model applicable to all cancer types in vivo. There is evidence, by stable isotope tracing and mass spectrometry, that some types of lung, liver and brain tumors display high levels of complete glucose oxidation in vivo [33]. Therefore, these studies suggest that, in vivo, the origin of the tumor and the environmental niche as well as the metastatic niche play a crucial role in shaping the metabolic profile of the tumor cells, challenging the Warburg effect model to be of general validity.
Even further complicating the metabolic routes in cancers, in the reverse Warburg model [34], cancer cells highjack CAFs and reprogram their metabolism to adopt an aerobic glycolysis, which is in part triggered by the increasing production of reactive oxygen species (ROS) by neighboring cancer cells. CAFs consequently secrete metabolites such as pyruvate and lactate, which are taken up by cancer cells and used to support their metabolic needs as alternative carbon sources. In this phenomenon mono-carboxylate transporters (MCTs), both on CAFs and cancer cells, facilitate the metabolite exchange between the two cells types [35]. CAFs might thus become continuously exhausted and replaced by novel recruited fibroblasts. This might explain the rapid loss of the human tumor stromal CAFs and their replacement by their murine counterparts in patient derived xenograft models [36].
CAFs also have the ability to reshape and change the extracellular matrix. In cancer, it is well-established that during cancer progression tissue fibrosis and increased ECM stiffness can appear due to changes in the stroma cell phenotype. The interlinked relationships between ECM remodeling, CAF and cancer progression were demonstrated by Bertero et al. in 2019 [37]. The authors found that stiffening of the ECM by CAFs activated the Yes-associated protein 1 (YAP) and Transcriptional coactivator with PDZ-binding motif (TAZ) transcriptional programs, increasing transcription of glutaminase synthase (GLS), lactate dehydrogenase A (LDHA) and aspartate/glutamate transporter SLC1A3 genes. These changes increased metabolite exchange that mutualistically sustained pro-tumor activities in both the cancer cells and the CAF compartment [37]. In hepatocellular carcinoma, induced ECM stiffness activated YAP downstream of c-Jun N-terminal kinases (JNK) and p38. In turn YAP activation increased the glycolytic pathway and migration of cancer cells [38]. YAP/TAZ has also been found to promote cancer cell proliferation by upregulating deoxynucleotide synthesis and inhibiting RAS-induced senescence in cells. However, YAP was inhibited by plating cells on soft substrates which induced expression of senescence markers [39]. These changes can also affect metabolism in cancer cells directly. Plating triple-negative breast cancer cells on denser collagen substrates has been shown to cause a shift towards aerobic glycolysis [40]. In a mechanistically more detailed study, Park et al. in 2019 demonstrated that F-actin regulated degradation of phosphofructokinase (PFK) via ligase tripartite motif containing-21 (TRIM21) [41]. Under mechanical stress TRIM21 was bound to the F-actin bundles of the cytoskeleton and thus PFK degradation was prevented. Transformed cells bypassed this form of regulation by having thick bundles of F-actin that did not respond to mechanical cues. These data indicate that ECM remodeling is directly influenced by CAFs and reciprocally also influences CAFs in cancer. Moreover, these changes in ECM influence directly or indirectly the metabolic phenotype and progression of tumors.
2.2. Amino Acid Metabolism
Human proteins are composed by 20 different proteinogenic amino acids, which are divided into two different groups: the essential amino acids (EAAs: histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan and valine) and the non-essential amino acids (NEAAs: alanine, aspartate, asparagine, arginine, cysteine, glutamate, glutamine, glycine, proline, serine and tyrosine) [42]. In cancer, amino acids, apart from their normal role as protein building blocks, can serve as alternative energy source to fuel the TCA cycle and can regulate redox status as well as antioxidant defense [42][43]. They can also act as substrates for post-translational and epigenetic transformation [42][43]. These versatile aspects of amino acids led to a surge of interest in further investigating their mechanistic roles in tumor metabolism.
One of the key amino acids, which is abundant in human plasma, is glutamine (Gln). Gln acts as an important precursor for the synthesis of proteins, nucleotides, fatty acids and other critical molecules [44][45]. There are extensive studies on the role of glutamine in regulating cancer cell metabolism [46][30][31]. Several of these have reported crucial roles of Gln metabolism in the interaction between CAFs and tumor cells (Table 1). In ovarian cancer, CAFs were shown to generate high levels of Gln by glutamine synthetase (GS). In this cancer, CAF-derived Gln was exported to the tumor cells and converted to glutamate by the enzyme glutaminase. This further supported tumor cell growth by anaplerosis (replenishment process of metabolic pathway intermediates) of the TCA [47]. Indeed, co-inhibition of glutamine synthetase (GLUL) in CAFs and glutaminase (GLS) in ovarian cancer cells abrogated cancer cell growth better than each individual treatment [47]. Furthermore, in a recent study, it was found that CAFs migrate from the glutamine depleted core of tumors towards more glutamine rich areas. This migration based on a glutamine gradient was dependent on protein kinase B (ATK2) and allowed tumor cells to escape the orginal tumor site [48].
Additionally, CAF-derived Gln has been reported to activate a process called reductive carboxylation in pancreatic ductal adenocarcinoma (PDAC) [49]. Reductive carboxylation of glutamine is a thoroughly studied metabolic route in which glutamine is converted to α-ketoglutarate (α-KG) to enter the TCA cycle. Then, in a reversal of the TCA cycle, α-KG is converted to citrate, which is finally exported to the cytoplasm where it contributes to fatty acid biosynthesis. This glutamine-dependent metabolic reprogramming was induced by the transfer of CAF-derived exosomes (CDEs containing glutamine) into PDAC cells and was shown to be independent of KRAS activation [49].
Further research suggests that CAFs can transfer aspartate to cancer cells via the SLC1A3 transporter (also known as excitatory amino acid transporter 1 [EAAT1]), to facilitate nucleotide synthesis in the tumor cells [37], whereas glutamine-derived glutamate from cancer cells is taken up by CAFs through the same transporter.
Pancreatic stellate cells (PSCs), one of the best studied CAF subtypes in PDAC, can be reprogrammed to secrete alanine through activation of autophagy. The secreted alanine is taken up by the tumor cells and converted to pyruvate which fuels the TCA. This further allows PDAC cells to divert glucose carbon atoms into serine/glycine metabolism for sustaining their proliferation. The dysregulation of tryptophan metabolism due to tumor–CAF interaction has also been reported (Table 1). Yet, there are still many amino acids which have not been explored in the metabolic crosstalk between CAF and tumor cells. Obviously, further research is needed to dissect the metabolic function of other amino acids within tumor–CAFs interactions.
2.3. Lipid Metabolism
Lipids are macromolecules that are soluble in non-polar solvents. In biology they include substances such as fats, waxes, oils, vitamins and steroid hormones. Fatty acids (FAs) are the building blocks of various lipids. Lipids and FAs have main functions in cells: (i) they build up the structural components of cell membranes which mainly consist of glycerophospholipids and sphingolipids (ii) they provide energy storage in the form of triglycerides and (iii) they work as signaling molecules [50]. Given these functions, it is not surprising that increased de novo FA synthesis has been observed in various types of cancer in order to support rapid tumor cell growth [51][52].
Limited studies have investigated how the dysregulation of lipid metabolism in CAFs affects tumorigenesis. In an early study in 2013 by Kamphorst et al. the authors were able to show that hypoxic cancer cells scavenge FA from lysophospholipids [53]. In 2019, Auciello et al. further demonstrated that CAFs secrete lysophospholipids, particularly lysophosphatidylcholine, to promote tumor proliferation and metastatic processes in PDAC [19]. In a similar study, lysophosphatidic acid from ovarian cancer cells induced a CAF phenotype in peritumoral fibroblasts, indicating again how the crosstalk between CAFs and cancer cells is mutualistic in nature [54]. Finally, a recent study indicates that lipids secreted by CAFs are taken up by colorectal cancer (CRC) cells and that deletion of the fatty acid synthase (FASN) or the inhibition of FA uptake in CAFs reduced CRC cell migration [55].
Recently, more attention has come to an understanding on how mechanical forces in the TME can regulate lipid metabolism. In 2019, Romani et al. demonstrated that a soft ECM microenvironment increased lipid and cholesterol synthesis [56]. Mechanistically it was identified that reduced mechanical stress on the Golgi apparatus led to sterol regulatory-element binding proteins (SREBP) activation and translocation to the nucleus, where it induced activation of lipid synthesis [56]. Conversely, in 2018 Boulter et al. uncovered that genetic deletion of the amino acid transporter and integrin coreceptor CD98hc (SLC3A2) indirectly influenced mechanosensing of integrins by impairment in sphingolipid metabolism [57].
2.4. Immune Modulation by CAF-Derived Metabolism
CAFs have been hypothesized to play an important role in immune evasion (for a more in depth review please refer to [58]). CAF-secreted metabolites, such as lactate, can actively participate in an immunosuppressive TME. Several studies have attempted to further clarify the role of CAF-derived metabolites in modulating anti-tumor immunity. In breast cancer, CAF-secreted kynurenine, a tryptophan metabolite, was found to promote E-cadherin/Aryl hydrocarbon receptor (AhR)/S-phase kinase-associated protein 2 (Skp2) complex, leading to E-cadherin degradation, which supported cancer cell invasion [59]. Indoleamine 2,3-dioxygenase (IDO), the key enzyme in the degradation process of tryptophan to kynurenine, was found to be expressed by CAFs. The STAT3-dependent release of prostaglandin E2 by cancer cells triggered the upregulation of IDO in CAFs. Moreover, the co-expression of IDO in stromal fibroblasts and cyclooxygenase (COX2) in breast tumors was correlated with poor patient survival and metastasis spreading. CAFs-derived IDO can also induce T cell anergy and inhibit CD8+ cytotoxic activity [60]. Similarly, the upregulation of galectin-1 in lung cancer cells induced CAFs to overexpress tryptophan 2,3-dioxygenase (TDO2) and enabled CAFs to secrete kynurenine [61]. This kynurenine/TDO2 signaling was found to promote cancer growth and invasion, while suppressing the differentiation of dendritic cells through the AKT/cAMP response element-binding protein (CREB)/ WNK Lysine Deficient Protein Kinase 1 (WNK1) axis. The production of arginase II (ARG 2) by CAFs was also reported to hamper anti-tumor T cell functions [62].
Moreover, a reciprocal interaction between CAFs and neutrophils has been described [63]. Activated neutrophils are high producers of ROS [64]. Using a lymphoma mouse model, a study showed that tumor cells educated CAFs to enhance the recruitment of CD11b+Ly6G+ neutrophils via the CCL2-CCR-2 axis and accelerated tumor growth [65]. Activated neutrophils can also stimulate the transformation of MSCs into highly FAP expressing CAFs and promote the metastasis of gastric cancer cells via IL6/STAT3 axis [66]. In hepatocellular carcinoma, α-SMA+ CAFs are found to produce IL-6 for recruiting neutrophils via the activation of STAT3 and c-Jun kinase-programmed cell death ligand 1 (STAT3-PDL1) [67]. This signaling cascade induces neutrophil apoptosis and fosters tumor progression. Immunosuppressive roles of CAFs on macrophages and CD8+ T cells have also been shown to be ROS-dependent [68][69][70]. CAFs can additionally lead to extensive reorganization of the mitochondrial metabolism in prostate cancer in a STIRT1/PGC-1a dependent manner [71]. This leads to extensive mitochondria-generated ROS (mtROS) generation that supports pro-invasive features in prostate cancer cells.
3. Metabolic Approaches to Study the Cross-Talks between CAFs and Tumor Cells
The proper analysis of metabolism in cell-cell interaction is dependent on specific analytic techniques to measure the metabolome, which represents the pool of metabolites from diverse array of metabolic activities [72][73]. Here we discuss the different methods that allow us deciphering the metabolic crosstalk between different cells, e.g., of CAFs and tumor cells.
3.1. Mass Spectrometry-Based Metabolomics
Generally, mass spectrometry (MS) is the leading technology used to identify metabolites and quantify their concentration [74]. It allows obtaining both quantitative and qualitative information about the intracellular or extracellular metabolome composition of a given biological sample [72]. MS is commonly coupled with gas chromatography (GC) or liquid chromatography (LC), to allow pre-separation of metabolites. GS tandem MS requires the derivatization of metabolites and their conversion to the air phase. This limits the number of molecules that can reliably be measured. GC-MS, however, is an ideal technique for analyzing non-polar, volatile and non-volatile molecules of small molecular weight [75]. Conversely, LS-MS is well-suited to analyze thermally unstable molecules and requires a less extensive extraction process. These metabolites are then loaded on the MS to identify and decode the metabolic profiles of cells or tissues. This kind of metabolite analysis can be used for a targeted (with pre-defined or selected metabolites targets) and a non-targeted metabolomics approach.
In the studies of CAF and tumor cell interactions, the use of LC-MS is more popular compared with GC-MS (Table 1). This is probably due to the broader coverage of metabolite read-outs and a simpler methodical setup in LC-MS compared to GC-MS. A few studies have also used superior versions of the conventional LC, namely ultra-performance liquid chromatography (UPLC) or high-performance liquid chromatography (HPLC) to assess metabolism in tumor–CAF interaction. Both allow the separation of smaller particle sizes (<2 µm for UPLC) and provide a higher speed, resolution, and sensitivity [76]. However, MS is mostly used to analyze in vitro cultures and ex vivo tumor tissues. More applications of those techniques are needed to further explore CAF and tumor metabolic interactions under in vitro and/or in vivo settings. In the future, they will be particularly useful in delineating the role of small extracellular vesicles, which are derived from CAFs, and are capable of driving tumor metabolic rewiring. Moreover, integrating the MS-measurements with other metabolic approaches such as stable isotope labelling and seahorse assays seems an ideal way to decipher CAF and tumor cell metabolic interactions.
3.2. Metabolic Flux Analysis
While measuring the level of cellular metabolites provides crucial information, it is equally important to determine the pathways that are linked to them and how they interact. On its own, a drop in the levels of a metabolite can either point towards the reduction of the synthesis or the increased consumption of this metabolite via another pathway. One of the metabolic techniques used to determine the pathways that contribute to metabolite secretion is known as ‘metabolic flux analysis’ (MFA). MFA uses nutrients, such as glucose, amino acids, or lipids, which are labelled with the stable isotopes 13C or 14N, as the basis for metabolites’ identification and quantification [77]. Mass spectrometry is commonly used to analyze the labeled samples. Primarily, 13C-MFA aims to produce a quantitative cellular metabolic map by assigning the values of fluxes to the reactions in the network and by using confidence interval for every predicted flux [78].
Although MFA has been used to elucidate the metabolic changes that take place in cancer cells and their microenvironment in vitro, it is equally important to perform 13C-MFA under in vivo settings, which has been addressed recently [76][79]. In fact, the variations in experimental set-up between different in vitro studies on CAF–tumor interaction has also raised concerns about the most reliable workflow to use. Thus, its usage for understanding CAF–tumor cell interactions is still limited (Table 1), particularly under the in vivo or ex vivo settings. However, in vivo flux analysis provides a better comprehensive view of the metabolism at the cellular and whole-organism level which can complement in vitro approaches [80][81]. In particular, it will help to understand the nutrient exchange between CAFs and the other components of the TME, such as immune cells or pericytes, which are known to impact tumorigenesis. Furthermore, by using 13C-MFA in vivo, it will be possible to understand the contribution of each metabolic pathway to the generation of CAF-secreted oncogenic metabolites in a physiological relevant context. Accordingly, the integration of MFA with other computational approaches will lead to a better understanding of the CAF–tumor interaction-driven cancer metabolic rewiring in the future.
3.3. Seahorse Extracellular Flux
The Agilent Seahorse XF Analyzer is a powerful tool that can offer a rapid insight into the metabolic state of the cells. Seahorse protocols use a combination of metabolic inhibitors to quantify the rate of glycolysis and/or OXPHOS, via the measurement of extracellular acidification rate (ECAR) and oxygen consumption rate (OCR). By understanding the cellular responses to metabolic drugs, the different respiratory phases can be defined and the cellular bioenergetics status of the cells can be inferred [82].
In ECAR measurement, cells are stimulated with 2,4-Dinitrophenol (2,4-DNP, an oxidative phosphorylation inhibitor) and 2-Deoxy-D-Glucose (2-DG, a hexokinase inhibitor). 2-4 DNP induces maximal glycolytic capacity, whereas 2-DG is a glucose analog that completely shuts glycolysis. For measuring OCR, cells are exposed to mitochondria perturbing reagents. Oligomycin, carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) and rotenone coupled with antimycin are added to the cells in a sequential manner to induce changes in mitochondrial activity. Oligomycin blocks the ATP synthase (complex V) and suppresses OCR. FCCP is a mitochondrial uncoupler which disrupts the proton gradient and mitochondrial membrane potential, and drives OCR to a maximal level, which is then reduced to a minimal level by antimycin A and rotenone (complex III and complex I inhibitor), finally disrupting mitochondrial respiration. The administration of these drugs at different time-points causes the fluctuation in the OCR level. These data can then be used to calculate basal respiration, ATP-linked respiration, proton leak, maximal respiration capacity, reserve capacity and non-mitochondrial respiration [83].
An indirect in vitro study set-up using the conditioned media, of either the tumor cells or the CAFs, is commonly used to study CAF–tumor cell interaction with Seahorse (Table 1). This is obviously due to the simplicity of such a set-up. In addition, most studies have only been performed on a 2D cell culture monolayer so far. However, it is commonly accepted that 3D cell cultures better recapitulate the physiological parameters of a tumor; they have been described to better mimic cell–cell interaction and environmental factors (gradient of oxygen and nutrient supply) compared to their 2D counterparts [84][85].
In a more advanced setup, Demircioglu et al., 2020 have used Seahorse to analyze isolated cells from ex vivo tumor samples [86]. Such an approach may give deeper insights into the physiological and metabolic state of a tumor. Yet, the prior tissue processing steps to isolate the cells need to be considered as these may significantly alter the initial tumor metabolic state.
Thus, further explorations on 3D cultures and direct co-culture settings with Seahorse, in combination with other metabolic measurements (such as stable isotope tracing and non-destructive metabolic imaging approaches), are definitively warranted in order to gain greater mechanistic insights into CAF–tumor cell interaction.
3.4. Computational Approaches to Unravel Tumor-CAF Metabolic Reprogramming
The increasing use of modern analytical techniques has helped characterizing tumor metabolism. However, the heterogeneity of the tumor metabolic rewiring is still not yet fully understood. Studies often only touch upon the phenotypic analysis without providing an in-depth mechanism. This is mostly due to the technical challenges and experimental limitations in the field of metabolomics. While metabolomics studies provide powerful insights into the cellular metabolic status of cells, the information regarding the cause or effect of metabolite changes is still missing, hence limiting our understanding of the underlying mechanisms. The cell’s metabolome constitutes the amplified and integrated signals from various levels, either at the transcriptional level or post-translational level [87]. Therefore, a multi-omics approach (proteomics, transcriptomics and metabolomics) may provide a bigger picture to identify the key molecular regulators of a complex disease, like cancer. Further integration of ‘omics’ data to build personalized in silico models (i.e., models which represent each patient individually) could shed light on the metabolic heterogeneity and the regulatory interactions between different classes of biomolecules (genes, proteins and metabolites), especially in CAF subpopulations which are continuously being identified.
The principle of network modelling is to present high-throughput data in a robust and predictive way [88]. Metabolic models are computational tools showing the metabolites as a set of nodes and the enzymatic conversion from one metabolite to another as edges [89]. The main advantage of computational metabolic networks is the possibility to narrow down the number of potential targets to be validated in vitro, therefore saving time and money. They also allow studying the complex interactions between the tumor and its microenvironment on multiple scales and generating patient-specific models [90]. As such, they represent important tools to improve drug testing, thereby making treatments more accurate and beneficial to the patients [91].
Genome-scale metabolic reconstructions, such as Recon2 [92], iHSA [93], or Human1 [94], consist of a library of all the possible reactions that can occur in an organism, as well as a set of genes that control the enzymes and transporters that allow the reactions to take place [95]. There are two types of metabolic models. Kinetic models rely on ordinary differential equations and kinetic rates. Their applicability is, however, limited, due to a high number of parameters to be determined [96]. Constraint-based models focus on steady states and do not integrate parameters, which allows reconstructing and analyzing larger models. Over the last few years, these models have gained interest to model the metabolic status of a cell [97]. One important advantage of constraint-based modelling is the possibility to integrate multi-omics data, such as transcriptomics [98], proteomics [99]or metabolomics [100], to constrain genome-scale metabolic reconstructions and generate context-specific models [101]. Several algorithms exist that build context-specific models such as iMAT [102], INIT [103], mCADRE [104] and the FASTCORE family [105][106][107]. Finally, in silico gene knock-out can be applied to identify potential drug targets. Gene knock-out consists of blocking all the reactions associated with a specific gene and assesses its effect on the objective function of the model, i.e., biomass production. The genes that significantly reduce growth are considered as essential and, therefore, as potential drug targets [108].
However, modelling a complex system such as the tumor microenvironment remains challenging, mainly due to the difficulty in modelling the interactions between multiple cell types. Indeed, such models already exist for microbial communities [109], however, similar approaches applied to multiple human cell types are still widely unexplored. In 2010, Lewis et al. built a constraint-based model for multiple cell interactions in the brain based on Recon1 and successfully simulated Alzheimer’s disease [110]. In 2015, Capuani et al. reconstructed a small, unconstrained model (75 reactions) for the lactate shuttle between CAFs and tumor cells [111]. More recently, in 2018 Shan et al. modeled the impact of the tumor microenvironment on the Warburg effect and glutamine addiction in cancer cells and found that the reverse Warburg effect provided growth advantage to the tumors originating from deep tissues [112]. Finally, in 2019 Damiani et al. developed a new algorithm (single-cell flux balance analysis) that allows integrating single-cell RNA-seq data into models of breast and lung cancer. This method allowed them to model an heterogeneous cancer cell population, to identify cancer cell subpopulations based on their growth rate and enabled to represent metabolite exchange between different cell types [36][113].
These first studies strongly demonstrate that there is no doubt that constrain-based modelling combined with single-cell sequencing will become one of the most important tools to study tumor–microenvironment interactions. Additionally, many efforts are now directed towards the integration of signaling data into metabolic models.
This entry is adapted from the peer-reviewed paper 10.3390/cells10020304
References
- Dhom, G. The cancer cell and the connective tissue: A historical review. Pathologe 1994, 15, 271–278.
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458.
- Dvorak, H.F. Tumors: Wounds that do not heal-redux. Cancer Immunol. Res. 2015, 3, 1–11.
- Xing, F.; Saidou, J.; Watabe, K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front. Biosci. 2010, 15, 166–179.
- Romero, I.L.; Mukherjee, A.; Kenny, H.A.; Litchfield, L.M.; Lengyel, E. Molecular pathways: Trafficking of metabolic resources in the tumor microenvironment. Clin. Cancer Res. 2015, 21, 680–686.
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. DMM Dis. Model. Mech. 2018, 11.
- Li, Z.; Zhang, J.; Zhou, J.; Lu, L.; Wang, H.; Zhang, G.; Wan, G.; Cai, S.; Du, J. Nodal Facilitates Differentiation of Fibroblasts to Cancer-Associated Fibroblasts that Support Tumor Growth in Melanoma and Colorectal Cancer. Cells 2019, 8, 538.
- Santolla, M.F.; Vivacqua, A.; Lappano, R.; Rigiracciolo, D.C.; Cirillo, F.; Galli, G.R.; Talia, M.; Brunetti, G.; Miglietta, A.M.; Belfiore, A.; et al. GPER Mediates a Feedforward FGF2/FGFR1 Paracrine Activation Coupling CAFs to Cancer Cells Toward Breast Tumor Progression. Cells 2019, 8, 223.
- Eiro, N.; Gonzalez, L.O.; Fraile, M.; Cid, S.; Schneider, J.; Vizoso, F.J. Breast cancer tumor stroma: Cellular components, phenotypic heterogeneity, intercellular communication, prognostic implications and therapeutic opportunities. Cancers 2019, 11, 664.
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186.
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115.
- Almeida-Porada, G.; Atala, A.J.; Porada, C.D. Therapeutic Mesenchymal Stromal Cells for Immunotherapy and for Gene and Drug Delivery. Mol. Ther. Methods Clin. Dev. 2020, 16, 204–224.
- Sage, E.K.; Thakrar, R.M.; Janes, S.M. Genetically modified mesenchymal stromal cells in cancer therapy. Cytotherapy 2016, 18, 1435–1445.
- Hmadcha, A.; Martin-Montalvo, A.; Gauthier, B.R.; Soria, B.; Capilla-Gonzalez, V. Therapeutic Potential of Mesenchymal Stem Cells for Cancer Therapy. Front. Bioeng. Biotechnol. 2020, 8, 1–13.
- Barrett, R.; Puré, E. Cancer-associated fibroblasts: Key determinants of tumor immunity and immunotherapy. Curr. Opin. Immunol. 2020, 64, 80–87.
- Salimifard, S.; Masjedi, A.; Hojjat-Farsangi, M.; Ghalamfarsa, G.; Irandoust, M.; Azizi, G.; Mohammadi, H.; Keramati, M.R.; Jadidi-Niaragh, F. Cancer associated fibroblasts as novel promising therapeutic targets in breast cancer. Pathol. Res. Pract. 2020, 216, 152915.
- Bhattacharya, S.D.; Mi, Z.; Talbot, L.J.; Guo, H.; Kuo, P.C. Human mesenchymal stem cell and epithelial hepatic carcinoma cell lines in admixture: Concurrent stimulation of cancer-associated fibroblasts and epithelial-to-mesenchymal transition markers. Surgery 2012, 152, 449–454.
- Hossen, M.N.; Rao, G.; Dey, A.; Robertson, J.D.; Bhattacharya, R.; Mukherjee, P. Gold Nanoparticle Transforms Activated Cancer-Associated Fibroblasts to Quiescence. ACS Appl. Mater. Interfaces 2019, 11, 26060–26068.
- Auciello, F.R.; Bulusu, V.; Oon, C.; Tait-Mulder, J.; Berry, M.; Bhattacharyya, S.; Tumanov, S.; Allen-Petersen, B.L.; Link, J.; Kendsersky, N.D.; et al. A stromal lysolipid–autotaxin signaling axis promotes pancreatic tumor progression. Cancer Discov. 2019, 9, 617–627.
- Alt, E.; Welte, G.; Li, J.; Hennessy, B.T.; Devarajan, E.; Krishnappa, S.; Pinilla, S.; Droll, L.; Jotzu, C. Adipose tissue-derived stem cells differentiate into carcinoma-associated fibroblast-like cells under the influence of tumor-derived factors. Anal. Cell. Pathol. 2010, 33, 61–79.
- McDonald, L.T.; LaRue, A.C. Hematopoietic stem cell derived carcinoma-associated fibroblasts: A novel origin. Int. J. Clin. Exp. Pathol. 2012, 5, 863–873.
- Augsten, M. Cancer-Associated Fibroblasts as Another Polarized Cell Type of the Tumor Microenvironment. Front. Oncol. 2014, 4, 1–8.
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. Il1-induced Jak/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019, 9, 282–301.
- Kieffer, Y.; Hocine, H.R.; Gentric, G.; Pelon, F.; Bernard, C.; Bourachot, B.; Lameiras, S.; Albergante, L.; Bonneau, C.; Guyard, A.; et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020, 10, 1330–1351.
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019, 9, 1102–1123.
- Mizutani, Y.; Kobayashi, H.; Iida, T.; Asai, N.; Masamune, A.; Hara, A.; Esaki, N.; Ushida, K.; Mii, S.; Shiraki, Y.; et al. Meflin-positive cancer-associated fibroblasts inhibit pancreatic carcinogenesis. Cancer Res. 2019, 79, 5367–5381.
- Reina-Campos, M.; Moscat, J.; Diaz-Meco, M. Metabolism shapes the tumor microenvironment. Curr. Opin. Cell Biol. 2017, 48, 47–53.
- Ocaña, M.C.; Martínez-Poveda, B.; Quesada, A.R.; Medina, M.Á. Metabolism within the tumor microenvironment and its implication on cancer progression: An ongoing therapeutic target. Med. Res. Rev. 2019, 39, 70–113.
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530.
- Fadaka, A.; Ajiboye, B.; Ojo, O.; Adewale, O.; Olayide, I.; Emuowhochere, R. Biology of glucose metabolization in cancer cells. J. Oncol. Sci. 2017, 3, 45–51.
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899.
- Courtney, K.D.; Bezwada, D.; Mashimo, T.; Pichumani, K.; Vemireddy, V.; Funk, A.M.; Wimberly, J.; McNeil, S.S.; Kapur, P.; Lotan, Y.; et al. Isotope Tracing of Human Clear Cell Renal Cell Carcinomas Demonstrates Suppressed Glucose Oxidation In Vivo. Cell Metab. 2018, 28, 793–800.e2.
- Sanderson, S.M.; Locasale, J.W. Revisiting the Warburg Effect: Some Tumors Hold Their Breath. Cell Metab. 2018, 28, 669–670.
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001.
- Knudsen, E.S.; Balaji, U.; Freinkman, E.; McCue, P.; Witkiewicz, A.K. Unique metabolic features of pancreatic cancer stroma: Relevance to the tumor compartment, prognosis, and invasive potential. Oncotarget 2016, 7, 78396–78411.
- Blomme, A.; Van Simaeys, G.; Doumont, G.; Costanza, B.; Bellier, J.; Otaka, Y.; Sherer, F.; Lovinfosse, P.; Boutry, S.; Palacios, A.P.; et al. Murine stroma adopts a human-like metabolic phenotype in the PDX model of colorectal cancer and liver metastases. Oncogene 2018, 37, 1237–1250.
- Bertero, T.; Oldham, W.M.; Grasset, E.M.; Bourget, I.; Boulter, E.; Pisano, S.; Hofman, P.; Bellvert, F.; Meneguzzi, G.; Bulavin, D.V.; et al. Tumor-Stroma Mechanics Coordinate Amino Acid Availability to Sustain Tumor Growth and Malignancy. Cell Metab. 2019, 29, 124–140.e10.
- Liu, Q.-P.; Luo, Q.; Deng, B.; Ju, Y.; Song, G.-B. Stiffer Matrix Accelerates Migration of Hepatocellular Carcinoma Cells through Enhanced Aerobic Glycolysis Via the MAPK-YAP Signaling. Cancers 2020, 12, 490.
- Santinon, G.; Brian, I.; Pocaterra, A.; Romani, P.; Franzolin, E.; Rampazzo, C.; Bicciato, S.; Dupont, S. d NTP metabolism links mechanical cues and YAP / TAZ to cell growth and oncogene-induced senescence. EMBO J. 2018, 37, 1–16.
- Mah, E.J.; Lefebvre, A.E.Y.T.; McGahey, G.E.; Yee, A.F.; Digman, M.A. Collagen density modulates triple-negative breast cancer cell metabolism through adhesion-mediated contractility. Sci. Rep. 2018, 8, 1–11.
- Park, J.S.; Burckhardt, C.J.; Lazcano, R.; Solis, L.M.; Isogai, T.; Li, L.; Chen, C.S.; Gao, B.; Minna, J.D.; Bachoo, R.; et al. Mechanical regulation of glycolysis via cytoskeleton architecture. Nature 2020, 578, 621–626.
- Choi, B.H.; Coloff, J.L. The diverse functions of non-essential amino acids in cancer. Cancers 2019, 11, 675.
- Lieu, E.L.; Nguyen, T.; Rhyne, S.; Kim, J. Amino acids in cancer. Exp. Mol. Med. 2020, 52, 15–30.
- Xie, H.; Simon, M.C. Oxygen availability and metabolic reprogramming in cancer. J. Biol. Chem. 2017, 292, 16825–16832.
- Sanford-Crane, H.; Abrego, J.; Sherman, M.H. Fibroblasts as modulators of local and systemic cancer metabolism. Cancers 2019, 11, 619.
- Lopes-Coelho, F.; André, S.; Félix, A.; Serpa, J. Breast cancer metabolic cross-talk: Fibroblasts are hubs and breast cancer cells are gatherers of lipids. Mol. Cell. Endocrinol. 2018, 462, 93–106.
- Yang, L.; Achreja, A.; Yeung, T.L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700.
- Mestre-Farrera, A.; Bruch-Oms, M.; Peña, R.; Rodríguez-Morató, J.; Alba-Castellón, L.; Comerma, L.; Quintela-Fandino, M.; Duñach, M.; Baulida, J.; Pozo, Ó.J.; et al. Glutamine-directed migration of cancer-activated fibroblasts facilitates epithelial tumor invasion. Cancer Res. 2020.
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016, 5.
- Ko, C.W.; Qu, J.; Black, D.D.; Tso, P. Regulation of intestinal lipid metabolism: Current concepts and relevance to disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 169–183.
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Lipid metabolism in cancer cells under metabolic stress. Br. J. Cancer 2019, 120, 1090–1098.
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22.
- Kamphorst, J.J.; Cross, J.R.; Fan, J.; De Stanchina, E.; Mathew, R.; White, E.P.; Thompson, C.B.; Rabinowitz, J.D. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. USA 2013, 110, 8882–8887.
- Radhakrishnan, R.; Ha, J.H.; Jayaraman, M.; Liu, J.; Moxley, K.M.; Isidoro, C.; Sood, A.K.; Song, Y.S.; Dhanasekaran, D.N. Ovarian cancer cell-derived lysophosphatidic acid induces glycolytic shift and cancer-associated fibroblast-phenotype in normal and peritumoral fibroblasts. Cancer Lett. 2019, 442, 464–474.
- Gong, J.; Lin, Y.; Zhang, H.; Liu, C.; Cheng, Z.; Yang, X.; Zhang, J.; Xiao, Y.; Sang, N.; Qian, X.; et al. Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis. 2020, 11.
- Romani, P.; Brian, I.; Santinon, G.; Pocaterra, A.; Audano, M.; Pedretti, S.; Mathieu, S.; Forcato, M.; Bicciato, S.; Manneville, J.B.; et al. Extracellular matrix mechanical cues regulate lipid metabolism through Lipin-1 and SREBP. Nat. Cell Biol. 2019, 21, 338–347.
- Boulter, E.; Estrach, S.; Tissot, F.S.; Hennrich, M.L.; Tosello, L.; Cailleteau, L.; de la Ballina, L.R.; Pisano, S.; Gavin, A.C.; Féral, C.C. Cell metabolism regulates integrin mechanosensing via an SLC3A2-dependent sphingolipid biosynthesis pathway. Nat. Commun. 2018, 9.
- De Jaeghere, E.A.; Denys, H.G.; De Wever, O. Fibroblasts Fuel Immune Escape in the Tumor Microenvironment. Trends Cancer 2019, 5, 704–723.
- Chen, J.Y.; Li, C.F.; Kuo, C.C.; Tsai, K.K.; Hou, M.F.; Hung, W.C. Cancer/stroma interplay via cyclooxygenase-2 and indoleamine 2,3-dioxygenase promotes breast cancer progression. Breast Cancer Res. 2014, 16, 1–14.
- Cheng, J.; Deng, Y.; Yi, H.; Wang, G.; Fu, B.; Chen, W.; Liu, W.; Tai, Y.; Peng, Y.; Zhang, Q. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis 2016, 5, e198.
- Hsu, Y.L.; Hung, J.Y.; Chiang, S.Y.; Jian, S.F.; Wu, C.Y.; Lin, Y.S.; Tsai, Y.M.; Chou, S.H.; Tsai, M.J.; Kuo, P.L. Lung cancer-derived galectin-1 contributes to cancer associated fibroblast-mediated cancer progression and immune suppression through TDO2/kynurenine axis. Oncotarget 2016, 7, 27584–27598.
- Ino, Y.; Yamazaki-Itoh, R.; Oguro, S.; Shimada, K.; Kosuge, T.; Zavada, J.; Kanai, Y.; Hiraoka, N. Arginase II Expressed in Cancer-Associated Fibroblasts Indicates Tissue Hypoxia and Predicts Poor Outcome in Patients with Pancreatic Cancer. PLoS ONE 2013, 8, e55146.
- Monteran, L.; Erez, N. The dark side of fibroblasts: Cancer-associated fibroblasts as mediators of immunosuppression in the tumor microenvironment. Front. Immunol. 2019, 10, 1835.
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil Elastase, Proteinase 3, and Cathepsin G as Therapeutic Targets in Human Diseases. Pharmacol. Rev. 2010, 62, 726–759.
- Ren, G.; Zhao, X.; Wang, Y.; Zhang, X.; Chen, X.; Xu, C.; Yuan, Z.R.; Roberts, A.I.; Zhang, L.; Zheng, B.; et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFα. Cell Stem Cell 2012, 11, 812–824.
- Zhu, Q.; Zhang, X.; Zhang, L.; Li, W.; Wu, H.; Yuan, X.; Mao, F.; Wang, M.; Zhu, W.; Qian, H.; et al. The IL-6-STAT3 axis mediates a reciprocal crosstalk between cancer-derived mesenchymal stem cells and neutrophils to synergistically prompt gastric cancer progression. Cell Death Dis. 2014, 5, e1295.
- Cheng, Y.; Li, H.; Deng, Y.; Tai, Y.; Zeng, K.; Zhang, Y.; Liu, W.; Zhang, Q.; Yang, Y. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 422.
- Zhang, A.; Qian, Y.; Ye, Z.; Chen, H.; Xie, H.; Zhou, L.; Shen, Y.; Zheng, S. Cancer-associated fibroblasts promote M2 polarization of macrophages in pancreatic ductal adenocarcinoma. Cancer Med. 2017, 6, 463–470.
- Xiang, H.; Ramil, C.P.; Hai, J.; Zhang, C.; Wang, H.; Watkins, A.A.; Afshar, R.; Georgiev, P.; Sze, M.A.; Song, X.S.; et al. Cancer-Associated Fibroblasts Promote Immunosuppression by Inducing ROS-Generating Monocytic MDSCs in Lung Squamous Cell Carcinoma. Cancer Immunol. Res. 2020, 8, 436–450.
- Sampson, N.; Brunner, E.; Weber, A.; Puhr, M.; Schäfer, G.; Szyndralewiez, C.; Klocker, H. Inhibition of Nox4-dependent ROS signaling attenuates prostate fibroblast activation and abrogates stromal-mediated protumorigenic interactions. Int. J. Cancer 2018, 143, 383–395.
- Ippolito, L.; Morandi, A.; Taddei, M.L.; Parri, M.; Comito, G.; Iscaro, A.; Raspollini, M.R.; Magherini, F.; Rapizzi, E.; Masquelier, J.; et al. Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene 2019, 38, 5339–5355.
- Kim, Y.M.; Heyman, H.M. Mass spectrometry-based metabolomics. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2018; Volume 1775, pp. 107–118.
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171.
- Jang, C.; Chen, L.; Rabinowitz, J.D. Metabolomics and Isotope Tracing. Cell 2018, 173, 822–837.
- Parri, M.; Ippolito, L.; Cirri, P.; Ramazzotti, M.; Chiarugi, P. Metabolic cell communication within tumour microenvironment: Models, methods and perspectives. Curr. Opin. Biotechnol. 2020, 63, 210–219.
- Lagziel, S.; Lee, W.D.; Shlomi, T. Studying metabolic flux adaptations in cancer through integrated experimental-computational approaches. BMC Biol. 2019, 17, 1–11.
- Bantscheff, M.; Schirle, M.; Sweetman, G.; Rick, J.; Kuster, B. Quantitative mass spectrometry in proteomics: A critical review. Anal. Bioanal. Chem. 2007, 389, 1017–1031.
- Antoniewicz, M.R. A guide to 13C metabolic flux analysis for the cancer biologist. Exp. Mol. Med. 2018, 50, 19.
- Faubert, B.; Deberardinis, R.J. Analyzing tumor metabolism in vivo. Annu. Rev. Cancer Biol. 2017, 1, 99–117.
- Elia, I.; Fendt, S.M. In vivo cancer metabolism is defined by the nutrient microenvironment. Transl. Cancer Res. 2016, 5, S1284–S1287.
- Fernández-García, J.; Altea-Manzano, P.; Pranzini, E.; Fendt, S.M. Stable Isotopes for Tracing Mammalian-Cell Metabolism In Vivo. Trends Biochem. Sci. 2020, 45, 185–201.
- Application Brief Agilent Technologies. Measuring Glycolysis and Oxidative Metabolism in Cancer Cells. Available online: https://www.agilent.com (accessed on 19 August 2020).
- Plitzko, B.; Loesgen, S. Measurement of Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR) in Culture Cells for Assessment of the Energy Metabolism. Bio-Protocol 2018, 8.
- Qureshi-Baig, K.; Ullmann, P.; Haan, S.; Letellier, E. Tumor-Initiating Cells: A criTICal review of isolation approaches and new challenges in targeting strategies. Mol. Cancer 2017, 16, 1–16.
- Russell, S.; Wojtkowiak, J.; Neilson, A.; Gillies, R.J. Metabolic Profiling of healthy and cancerous tissues in 2D and 3D. Sci. Rep. 2017, 7, 1–11.
- Demircioglu, F.; Wang, J.; Candido, J.; Costa, A.S.H.; Casado, P.; de Luxan Delgado, B.; Reynolds, L.E.; Gomez-Escudero, J.; Newport, E.; Rajeeve, V.; et al. Cancer associated fibroblast FAK regulates malignant cell metabolism. Nat. Commun. 2020, 11, 1290.
- Sauer, U. Metabolic networks in motion: 13C-based flux analysis. Mol. Syst. Biol. 2006, 2, 62.
- Ji, Z.; Yan, K.; Li, W.; Hu, H.; Zhu, X. Mathematical and Computational Modeling in Complex Biological Systems. BioMed Res. Int. 2017, 2017, 5958321.
- Alm, E.; Arkin, A.P. Biological networks. Curr. Opin. Struct. Biol. 2003, 13, 193–202.
- Hadjicharalambous, M.; Wijeratne, P.A.; Vavourakis, V. From tumour perfusion to drug delivery and clinical translation of in silico cancer models. Methods 2020.
- Werner, H.M.J.; Mills, G.B.; Ram, P.T. Cancer systems biology: A peek into the future of patient care? Nat. Rev. Clin. Oncol. 2014, 11, 167–176.
- Thiele, I.; Swainston, N.; Fleming, R.M.T.; Hoppe, A.; Sahoo, S.; Aurich, M.K.; Haraldsdottir, H.; Mo, M.L.; Rolfsson, O.; Stobbe, M.D.; et al. A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 2013, 31, 419–425.
- Blais, E.M.; Rawls, K.D.; Dougherty, B.V.; Li, Z.I.; Kolling, G.L.; Ye, P.; Wallqvist, A.; Papin, J.A. Reconciled rat and human metabolic networks for comparative toxicogenomics and biomarker predictions. Nat. Commun. 2017, 8.
- Robinson, J.L.; Kocabaş, P.; Wang, H.; Cholley, P.E.; Cook, D.; Nilsson, A.; Anton, M.; Ferreira, R.; Domenzain, I.; Billa, V.; et al. An atlas of human metabolism. Sci. Signal. 2020, 13, eaaz1482.
- Thiele, I.; Palsson, B. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121.
- Angione, C. Human Systems Biology and Metabolic Modelling: A Review-From Disease Metabolism to Precision Medicine. BioMed Res. Int. 2019, 2019, 1–16.
- Patil, K.R.; Åkesson, M.; Nielsen, J. Use of genome-scale microbial models for metabolic engineering. Curr. Opin. Biotechnol. 2004, 15, 64–69.
- Granata, I.; Troiano, E.; Sangiovanni, M.; Guarracino, M.R. Integration of transcriptomic data in a genome-scale metabolic model to investigate the link between obesity and breast cancer. BMC Bioinform. 2019, 20, 162.
- Schultz, A.; Mehta, S.; Hu, C.W.; Hoff, F.W.; Horton, T.M.; Kornblau, S.M.; Qutub, A.A. Identifying Cancer Specific Metabolic Signatures Using Constraint-Based Models. In Proceedings of the Biocomputing 2017, Kohala Coast, HI, USA, 4–8 January 2017; World Scientific: Singapore, 2017; Volume 22, pp. 485–496.
- Aurich, M.K.; Fleming, R.M.T.; Thiele, I. MetaboTools: A comprehensive toolbox for analysis of genome-scale metabolic models. Front. Physiol. 2016, 7.
- Wegrzyn, A.B.; Herzog, K.; Gerding, A.; Kwiatkowski, M.; Wolters, J.C.; Dolga, A.M.; van Lint, A.E.M.; Wanders, R.J.A.; Waterham, H.R.; Bakker, B.M. Fibroblast-specific genome-scale modelling predicts an imbalance in amino acid metabolism in Refsum disease. FEBS J. 2020, 287, 5096–5113.
- Zur, H.; Ruppin, E.; Shlomi, T. iMAT: An integrative metabolic analysis tool. Bioinformatics 2010, 26, 3140–3142.
- Agren, R.; Bordel, S.; Mardinoglu, A.; Pornputtapong, N.; Nookaew, I.; Nielsen, J. Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using INIT. PLoS Comput. Biol. 2012, 8.
- Wang, Y.; Eddy, J.A.; Price, N.D. Reconstruction of genome-scale metabolic models for 126 human tissues using mCADRE. BMC Syst. Biol. 2012, 6, 153.
- Vlassis, N.; Pacheco, M.P.; Sauter, T. Fast Reconstruction of Compact Context-Specific Metabolic Network Models. PLoS Comput. Biol. 2014, 10, e1003424.
- Pacheco, M.P.; John, E.; Kaoma, T.; Heinäniemi, M.; Nicot, N.; Vallar, L.; Bueb, J.L.; Sinkkonen, L.; Sauter, T. Integrated metabolic modelling reveals cell-type specific epigenetic control points of the macrophage metabolic network. BMC Genom. 2015, 16, 809.
- Pacheco, M.P.; Bintener, T.; Ternes, D.; Kulms, D.; Haan, S.; Letellier, E.; Sauter, T. Identifying and targeting cancer-specific metabolism with network-based drug target prediction. EBioMedicine 2019, 43, 98–106.
- Bintener, T.; Pacheco, M.P.; Sauter, T. Towards the routine use of in silico screenings for drug discovery using metabolic modelling. Biochem. Soc. Trans. 2020, 48, 955–969.
- Heinken, A.; Thiele, I. Systems biology of host-microbe metabolomics. Wiley Interdiscip. Rev. Syst. Biol. Med. 2015, 7, 195–219.
- Lewis, N.E.; Schramm, G.; Bordbar, A.; Schellenberger, J.; Andersen, M.P.; Cheng, J.K.; Patel, N.; Yee, A.; Lewis, R.A.; Eils, R.; et al. Large-scale in silico modeling of metabolic interactions between cell types in the human brain. Nat. Biotechnol. 2010, 28, 1279–1285.
- Capuani, F.; De Martino, D.; Marinari, E.; De Martino, A. Quantitative constraint-based computational model of tumor-to-stroma coupling via lactate shuttle. Sci. Rep. 2015, 5, 11880.
- Shan, M.; Dai, D.; Vudem, A.; Varner, J.D.; Stroock, A.D. Multi-scale computational study of the Warburg effect, reverse Warburg effect and glutamine addiction in solid tumors. PLoS Comput. Biol. 2018, 14, e1006584.
- Damiani, C.; Maspero, D.; Di Filippo, M.; Colombo, R.; Pescini, D.; Graudenzi, A.; Westerhoff, H.V.; Alberghina, L.; Vanoni, M.; Mauri, G. Integration of single-cell RNA-seq data into population models to characterize cancer metabolism. PLoS Comput. Biol. 2019, 15, e1006733.