Non-alcoholic fatty liver disease (NAFLD) is a multifactorial disease in which environmental and genetic factors are involved. Although the molecular mechanisms involved in NAFLD onset and progression are not completely understood, the gut microbiome (GM) is thought to play a key role in the process, influencing multiple physiological functions. GM alterations in diversity and composition directly impact disease states with an inflammatory course, such as non-alcoholic steatohepatitis (NASH). However, how the GM influences liver disease susceptibility is largely unknown. Similarly, the impact of strategies targeting the GM for the treatment of NASH remains to be evaluated. This review provides a broad insight into the role of gut microbiota in NASH pathogenesis, as a diagnostic tool, and as a therapeutic target in this liver disease. We highlight the idea that the balance in metabolic fermentations can be key in maintaining liver homeostasis. We propose that an overabundance of alcohol-fermentation pathways in the GM may outcompete healthier, acid-producing members of the microbiota. In this way, GM ecology may precipitate a self-sustaining vicious cycle, boosting liver disease progression.
- NAFLD
- gut microbiome
- microbial metabolic pathway
- microbiome-based signature
- fecal microbiota transplantation
Accumulated evidence indicates that the GM interacts with the liver via the so-called the “liver–gut axis” [16–18]. Dysfunction of this axis, including gut microbial imbalances and mucosa permeability alterations, leads to the passage of metabolic byproducts of bacterial metabolism as well as microbial components to the portal system reaching the liver. The microbial components, called pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharide and peptidoglycan, are capable of inducing inflammatory responses mediated by the activation of pattern recognition receptors (PRRs), like Toll-like receptor (TLR), in Kupffer cells and hepatic stellate cells, leading to liver injury and fibrosis [19–21]. Besides, some metabolic byproducts of bacterial metabolism may interfere with glucose and lipid metabolism, as discussed below, contributing to the exacerbation of liver disease [22,23]. On the other hand, it is well known that GM and bile acids (BAs) closely interact and modulate each other; BAs prevent intestinal bacterial overgrowth and subsequent gut barrier dysfunction, and the GM regulates bile acid composition [24]. Given that BAs modulate host metabolism and immunity, through farnesoid X receptor (FXR) and membrane-associated G protein-coupled receptor (TGR5) signaling, an imbalance in gut bacteria and BAs may trigger metabolic diseases, such as NAFLD [25] (Figure 1).
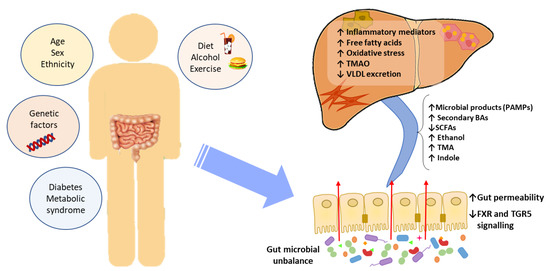
Figure 1. The effect of gut microbial unbalance in NAFLD. Different factors affect the gut microbiome. Gut microbial unbalance causes an increase in secondary BAs, which modulates FXR and FGR5 signaling, affecting the glucose and lipid metabolism and anti-inflammatory immune response. Besides, the increase in certain microbial metabolites mediates weakening of intestinal tight junction, enabling passage to the systemic circulation of PAMPs and microbial metabolites (such as ethanol) that reach the liver inducing inflammatory responses, liver injury and fibrosis. BAs, bile acids; FXR, farnesoid X receptor; PAMPs, pathogen associated molecular patterns; SCFAs, short-chain fatty acids; TGR5, membrane-associated G protein-coupled receptor; TMA, trimethylamine; TMAO, trimethylamine N-oxide; VLDL, very low-density lipoprotein.
Specific GM alterations have been correlated with the development and progression of NAFLD, both in human and in experimental animal models [6,22,26–28]. NAFLD patients exhibit more Gram-negative and fewer Gram-positive bacteria compared to healthy subjects, and disease progression correlates with phylum-level changes, such as an increase in Proteobacteria and a decrease in Firmicutes [29]. At the genus level, a significant increase in the abundance of Bacteroides and a decrease in Prevotella was observed in NASH patients, when compared to NAFLD patients without NASH [30]. Increased abundance of Ruminococcus in patients with fibrosis was also reported, as well as a relative increase in Streptococcus in obese patients with NAFLD [31]. These alterations are specifically linked to hepatic conditions, and are not the byproduct of insulin resistance, as demonstrated by Da Silva et al. [32]. However, although results point to a correlation between the GM and liver condition, the particular bacterial species involved are largely discordant across individual studies. These inconsistent results may be attributed to the lack of control and regularization for factors known to severely impact the GM, such as weight, diet, and drug intake [33,34]. Additionally, restricting the analysis to changes in diversity indices or comparisons at the phylum and other high-rank taxonomic levels is unlikely to yield insight into the molecular mechanisms involved. For these reasons, we are in need of approaches able to infer causal links, rather than mere statistical associations between specific bacterial species and liver conditions. The different methods for characterization of the GM are shown in Figure 2.
Meta-taxonomical approaches, if merely understood as the analysis of 16S sequences, are probably insufficient to unravel the causal links between GM composition and diseased states. Bacterial species contain pleomorphic genomes, with a conserved genetic core conserved among all members of a species, but also an accessory part that is highly variable among individual clones. The accessory part of the genome is often encoded in plasmids and other mobile genetic elements, thus subject to frequent change [35]. Due to this intrinsic genomic plasticity, strains of the same species frequently display significant phenotypic differences. Alternate metabolic profiles and even distinct virulence levels are common among strains of the same species, as exemplified by pathogenic and commensal E.coli [36–38]. As a consequence, meta-taxonomy alone may be unable to discriminate between strains that promote hepatic damage from those that do not. Similarly, if hepatic damage is the by-product of bacterial metabolism, it is likely that strains from different species produce similar hepatotoxic compounds as, in many species, non-essential, adaptive metabolic pathways are often encoded in mobile genetic elements [39,40].
Although the specific bacterial strains and species involved in NAFLD are still unknown, there is ample evidence that GM perturbations have a causal role in the development of the disease, rather than being a mere consequence of it. In animal models, it was shown that introducing a conventional GM in axenic mice increased monosaccharide absorption and triggered liver lipogenesis [41]. Similarly, faecal transplants from human donors with hepatic steatosis triggered a rapid development of hepatic steatosis in mice [42]. These phenomena suggest that GM does affect the host energy metabolism and fat storage. It may be thus key in the systemic inflammation associated with obesity, which leads to insulin resistance and hepatic steatosis. The molecular mechanisms by which GM alterations translate into hepatic damage are uncertain. However, several studies identified microbial metabolites associated with NAFLD, suggesting a role of certain gut-microbiome-derived metabolites in the pathogenesis and progression of NAFLD [42–45].
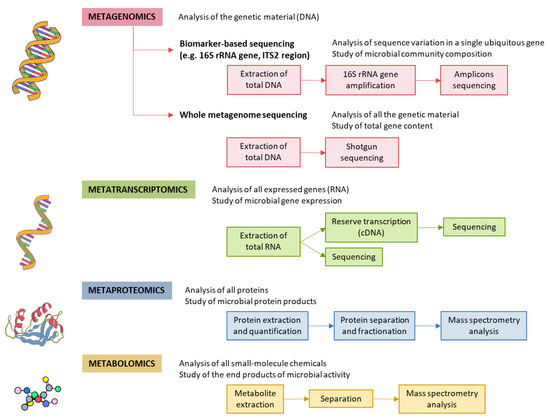
Figure 2. Methods for characterizing gut microbiota. 16S rRNA is highly conserved among bacterial species, except that it contains hypervariable regions that confer phylogenetic association; thus, 16S rRNA gene sequencing is widely used for phylogenetic reconstruction and quantification of microbial diversity. However, this technique does not make it possible to decipher functional changes in the microbiome or to find out the true impact of gut microbes on disease states. For this reason, several -omics approaches were put forward. These methods dig into genes for genetic information storage, transcription for gene expression, proteins for structural and metabolic activities, and metabolites for end products of metabolism. cDNA, complementary DNA; ITS2, internal transcribed spacer 2; rRNA, ribosomal RNA.
This entry is adapted from the peer-reviewed paper 10.3390/jcm9051369