O-GlcNAcylation is a posttranslational modification that occurs at serine and threonine residues of protein substrates by the addition of O-linked β-d-N-acetylglucosamine (GlcNAc) moiety. Two enzymes are involved in this modification: O-GlcNac transferase (OGT), which attaches the GlcNAc residue to the protein substrate, and O-GlcNAcase (OGA), which removes it. This biological balance is important for many biological processes, such as protein expression, cell apoptosis, and regulation of enzyme activity.
- OGT
- GlcNAcylation
- O-GlcNAc Transferase
- bioassay
- OGT inhibitor
1. Introduction
The study of O-GlcNAcylation, i.e., the reversible enzymatic posttranslational attachment/detachment of the N-acetylglucosamine sugar moiety to proteins, is a challenging topic in biochemistry and medicinal chemistry. Interestingly, this modification is regulated by only one pair of enzymes: O-GlcNAc transferase (OGT), which catalyses the post-translational transfer of N-acetylglucosamine via O-glycosidic linkage to the serine and threonine residues of proteins, and O-GlcNAcase (OGA), which can remove them (Figure 1) [1][2][3]. This topic has received a major boost in the last two decades with the discovery of small molecule inhibitors that modulate the process. Many studies have revealed the ubiquitous role of O-GlcNAcylation in biological processes, such as gene expression [3][4], cell cycle control [4][5], and regulation of enzyme activity [6][7][8][9]. Moreover, OGT competes with kinases for the modification site, leading to competition and cross-regulation of these translational modifications [9][10][11]. Dysfunction in the O-GlcNAcylation pathway has been associated with various chronic diseases [9][12][13]. In diabetes, it is associated with cognitive complications [12], while aberrant O-GlcNAcylation of tau protein in the brain leads to possible early onset of Alzheimer’s disease [13]. In cancer, O-GlcNAcylation appears to regulate tumour spread through adhesion and proliferation of cancer cells [1][14][15].
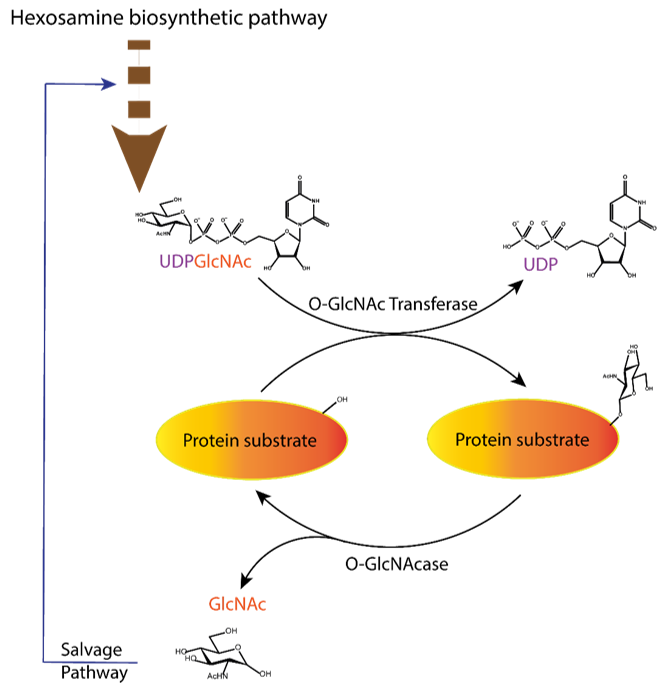
Figure 1. Dynamic equilibrium of the O-GlcNAcylation process. UDP-GlcNAc with the sugar moiety attached via α-O-glycosidic bond to the UDP is produced in the hexosamine biosynthetic pathway and subsequently attached to serine/threonine of protein substrates by O-GlcNAc transferase (OGT) via O-β-glycosidic bond. It is removed by O-GlcNAcase (OGA), and the GlcNAc formed after the OGA reaction re-enters the hexosamine pathway.
To study the mechanism of O-GlcNAcylation and the consequences of O-GlcNAcylated proteins in these diseases, molecular tools are needed to control the level of O-GlcNAcylation, i.e., inhibitors of both enzymes that modulate the O-GlcNAcylation status of proteins. In the case of OGA, many potent inhibitors have already been developed to control tauopathy levels in the brain. Two of them are currently in clinical trials for the treatment of Alzheimer’s disease and progressive supranuclear palsy (PSP), two diseases associated with tauopathy levels: MK-8719, which has passed preclinical trial tests, and ASN120290, which is scheduled for phase 2 clinical trials [16][17][18]. In contrast, the development of OGT inhibitors is less advanced and still in its infancy. As a transferase, OGT binds both the cofactor UDP-GlcNAc and a substrate protein. This provides additional opportunities, as blocking either of the two substrates should inhibit the enzyme. The goal of blocking both substrate and cofactor binding sites would likely result in inhibitors with molecular mass and physicochemical properties that would make these inhibitors non-drug-like. Moreover, as evident from some studies, the lack of stability of the isolated enzyme is a problem that complicates the search for potent inhibitors. It makes protein expression and purification a tedious practice, making in vitro assays with OGT problematic even on a routine basis [19].
Several strategies for designing OGT inhibitors have been described in the literature. We and others have reported bisubstrate/conjugate inhibitors consisting of a UDP mimic and a peptide fragment such as Goblin 1 or F20 [20][21]. OGT inhibitors based on UDP-GlcNAc mimics, such as UDP-5S-GlcNAc [22], have also been reported. Next, there are the UDP-mimics of the OSMI family, with OSMI-4 as the most potent binder to date with a Kd of 8 nM (Figure 2) [23][24][25]. To test new inhibitors, specific tools are needed, especially bioassays that can give a rapid and direct indication of the OGT inhibitory activity and the binding properties of the OGT inhibitor. Nowadays, with the development of new optical methods and the wider availability of high-throughput screening methods, there are many ways to design bioassays tailored to a specific macromolecular target. Somehow, this does not seem to be the case for the development of assays to assess the enzymatic activity of OGT and/or inhibitory potency of OGT inhibitors. The first reason is the instability of OGT [19]. As a result, despite many attempts, none of the published assays became a reference standard for the field. In the review (https://www.mdpi.com/142-3049/26/4/10370), we provide an overview of bioassays reported in the recent literature for testing OGT activity. We also highlight the most suitable assays and point out the new bioassays for screening OGT inhibitors (Figure 3). The first part of this review covers assays for measuring enzyme inhibition, kinetics, and different ways to follow the enzymatic response. The final part discusses assays that measure the binding affinity of the OGT inhibitors.
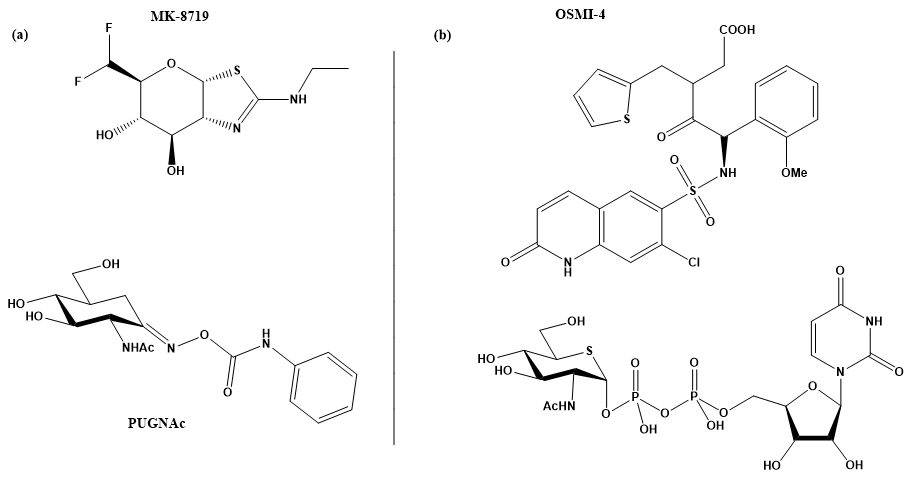
Figure 2. (a) MK-871914 and PUGNAc20 OGA inhibitors; MK-8719 potent clinical candidate as OGA inhibitor; (b) OSMI-4, the most potent OGT inhibitor to date, and UDP-5S-GlcNAc, an inhibitor biosynthesised by hijacking the hexosamine pathway using a per-acetylated 5S-GlcNAc.
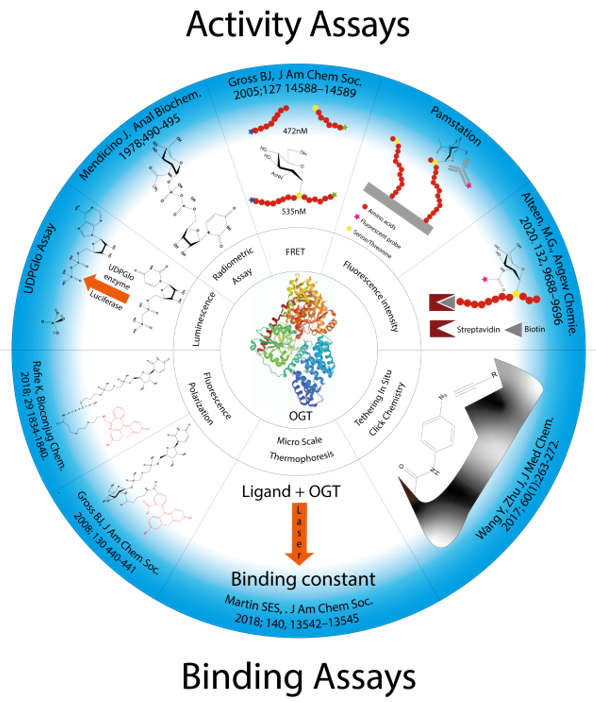
Figure 3. Presentation of all methods reported in the literature for screening OGT inhibitors and the techniques on which they are based.
This entry is adapted from the peer-reviewed paper 10.3390/molecules26041037
References
- Ferrer, C.M.; Lynch, T.P.; Sodi, V.L.; Falcone, J.N.; Schwab, L.P.; Peacock, D.L.; Vocadlo, D.J.; Seagroves, T.N.; Reginato, M.J. O-GlcNAcylation Regulates Cancer Metabolism and Survival Stress Signaling via Regulation of the HIF-1 Pathway. Cell 2014, 54, 820–831, doi:10.1016/j.molcel.2014.04.026.
- Aquino-Gil, M.; Pierce, A.; Perez-Cervera, Y.; Zenteno, E.; Lefebvre, T. OGT: a short overview of an enzyme standing out from usual glycosyltransferases. Soc. Trans. 2017, 45, 365–370, doi:10.1042/bst20160404.
- Özcan, S.; Andrali, S.S.; Cantrell, J.E. Modulation of transcription factor function by O-GlcNAc modification. et Biophys. Acta (BBA) - Bioenerg. 2010, 1799, 353–364, doi:10.1016/j.bbagrm.2010.02.005.
- Hart, G.W. Nutrient regulation of signaling and transcription. Biol. Chem. 2019, 294, 2211–2231, doi:10.1074/jbc.aw119.003226.
- Zhang, S.; Roche, K.; Nasheuer, H.P.; Lowndes, N.F. Modification of Histones by Sugar β-N-Acetylglucosamine (GlcNAc) Occurs on Multiple Residues, Including Histone H3 Serine 10, and Is Cell Cycle-regulated. Biol. Chem. 2011, 286, 37483–37495, doi:10.1074/jbc.m111.284885.
- Ma, J.; Wu, C.; Hart, G.W. Analytical and Biochemical Perspectives of Protein O-GlcNAcylation. Rev. 2021, 121, 1513–1581, doi:10.1021/acs.chemrev.0c00884.
- Fong, J.J.; Nguyen, B.L.; Bridger, R.; Medrano, E.E.; Wells, L.; Pan, S.; Sifers, R.N. β-N-Acetylglucosamine (O-GlcNAc) Is a Novel Regulator of Mitosis-specific Phosphorylations on Histone H3*. Biol. Chem. 2012, 287, 12195–12203, doi:10.1074/jbc.m111.315804.
- Harwood, K.R.; Hanover, J.A. Nutrient-driven O-GlcNAc cycling - think globally but act locally. Cell Sci. 2014, 127, 1857–1867, doi:10.1242/jcs.113233.
- Hart, G.W.; Slawson, C.; Ramirez-Correa, G.; Lagerlof, O. Cross Talk Between O-GlcNAcylation and Phosphorylation: Roles in Signaling, Transcription, and Chronic Disease. Annu Rev. Biochem. 2011, 7, 825–858, doi:10.1146/annurev-biochem-060608-102511.Cross.
- Wang, Z.; Udeshi, N.D.; Slawson, C.; Compton, P.D.; Sakabe, K.; Cheung, W.D.; Shabanowitz, J.; Hunt, N.F.; Hart, G.W. Extensive Crosstalk Between O-GlcNAcylation and Phosphorylation Regulates Cytokinesis. Signal. 2010, 3, ra2, doi:10.1126/scisignal.2000526.
- Kakade, P.S.; Budnar, S.; Kalraiya, R.D.; Vaidya, M.M. Functional Implications of O-GlcNAcylation-dependent Phosphorylation at a Proximal Site on Keratin 18. Biol. Chem. 2016, 291, 12003–12013, doi:10.1074/jbc.m116.728717.
- Bond, M.R.; Hanover, J.A. O-GlcNAc Cycling: A Link Between Metabolism and Chronic Disease. Rev. Nutr. 2013, 33, 205–229, doi:10.1146/annurev-nutr-071812-161240.
- Zhu, Y.; Shan, X.; Yuzwa, S.A.; Vocadlo, D.J. The Emerging Link between O-GlcNAc and Alzheimer Disease. Biol. Chem. 2014, 289, 34472–34481, doi:10.1074/jbc.r114.601351.
- Barkovskaya, A.; Seip, K.; Hilmarsdottir, B.; Maelandsmo, G.M.; Moestue, S.A.; Itkonen, H.M. O-GlcNAc Transferase Inhibition Differentially Affects Breast Cancer Subtypes. Rep. 2019, 9, 1–11, doi:10.1038/s41598-019-42153-6.
- Itkonen, H.M.; Urbanucci, A.; Martin, S.E.; Khan, A.; Mathelier, A.; Thiede, B.; Walker, S.; Mills, I.G. High OGT activity is essential for MYC-driven proliferation of prostate cancer cells. Theranostics 2019, 9, 2183–2197, doi:10.7150/thno.30834.
- Selnick, H.G.; Hess, J.F.; Tang, C.; Liu, K.; Schachter, J.B.; Ballard, J.E.; Marcus, J.N.; Klein, D.J.; Wang, X.; Pearson, M.; et al. Discovery of MK-8719, a Potent O-GlcNAcase Inhibitor as a Potential Treatment for Tauopathies. Med. Chem. 2019, 62, 10062–10097, doi:10.1021/acs.jmedchem.9b01090.
- Lee, H.E.; Lim, D.; Lee, J.Y.; Lim, S.M.; Pae, A.N. Recent tau-targeted clinical strategies for the treatment of Alzheimer ’ s disease. Future Sci. 2019, 11, 1845–1848.
- Shoeibi, A.; Olfati, N.; Litvan, I. Preclinical, phase I, and phase II investigational clinical trials for treatment of progressive supranuclear palsy. Expert Opin. Investig. Drugs 2018, 27, 349–361, doi:10.1080/13543784.2018.1460356.
- Kim, E.J.; Abramowitz, L.K.; Bond, M.R.; Love, D.C.; Kang, D.W.; Leucke, H.F.; Kang, D.W.; Ahn, J.-S.; Hanover, J.A. Versatile O-GlcNAc Transferase Assay for High-Throughput Identification of Enzyme Variants, Substrates, and Inhibitors. Bioconjugate Chem. 2014, 25, 1025–1030, doi:10.1021/bc5001774.
- Borodkin, V.S.; Schimpl, M.; Gundogdu, M.; Rafie, K.; Dorfmueller, H.C.; Robinson, D.A.; Van Aalten, D.M.F. Bisubstrate UDP–peptide conjugates as human O-GlcNAc transferase inhibitors. J. 2014, 457, 497–502, doi:10.1042/bj20131272.
- Zhang, H.; Tomašič, T.; Shi, J.; Weiss, M.; Ruijtenbeek, R.; Anderluh, M.; Pieters, R.J. Inhibition of O-GlcNAc transferase (OGT) by peptidic hybrids. MedChemComm 2018, 9, 883–887, doi:10.1039/c8md00115d.
- Liu, T.-W.; Zandberg, W.F.; Gloster, T.M.; Deng, L.; Murray, K.D.; Shan, X.; Vocadlo, D.J. Metabolic Inhibitors of O‐GlcNAc Transferase That Act In Vivo Implicate Decreased O‐GlcNAc Levels in Leptin‐Mediated Nutrient Sensing. Chem. Int. Ed. 2018, 57, 7644–7648, doi:10.1002/anie.201803254.
- Ortiz-Meoz, R.F.; Jiang, J.; Lazarus, M.B.; Orman, M.; Janetzko, J.; Fan, C.; Duveau, D.Y.; Tan, Z.-W.; Thomas, C.J.; Walker, S. A Small Molecule That Inhibits OGT Activity in Cells. ACS Chem. Biol. 2015, 10, 1392–1397, doi:10.1021/acschembio.5b00004.
- Gross, ‡ B.J.; Kraybill, † A.B.C.; Walker, † S. Discovery ofO-GlcNAc Transferase Inhibitors. Am. Chem. Soc. 2005, 127, 14588–14589, doi:10.1021/ja0555217.
- Martin, S.E.S.; Tan, Z.-W.; Itkonen, H.M.; Duveau, D.Y.; Paulo, J.A.; Janetzko, J.; Boutz, P.L.; Törk, L.; Moss, F.A.; Thomas, C.J.; et al. Structure-Based Evolution of Low Nanomolar O-GlcNAc Transferase Inhibitors. Am. Chem. Soc. 2018, 140, 13542–13545, doi:10.1021/jacs.8b07328.