Inherited retinal dystrophies (IRDs) are a group of retinal disorders that cause progressive and severe loss of vision because of retinal cell death, mainly photoreceptor cells. IRDs include retinitis pigmentosa (RP), the most common IRD. IRDs present a genetic and clinical heterogeneity that makes it difficult to achieve proper treatment. The progression of IRDs is influenced, among other factors, by the activation of the immune cells (microglia, macrophages, etc.) and the release of inflammatory molecules such as chemokines and cytokines. Upregulation of tumor necrosis factor alpha (TNFα), a pro-inflammatory cytokine, is found in IRDs. This cytokine may influence photoreceptor cell death. Different cell death mechanisms are proposed, including apoptosis, necroptosis, pyroptosis, autophagy, excessive activation of calpains, or parthanatos for photoreceptor cell death. Some of these cell death mechanisms are linked to TNFα upregulation and inflammation. Therapeutic approaches that reduce retinal inflammation have emerged as useful therapies for slowing down the progression of IRDs.
1. Introduction
Inherited retinal dystrophies (IRDs) constitute a heterogeneous group of retinal diseases that cause the progressive loss of vision. IRDs affect about 1 in 2000 to 3000 individuals [
1]. IRDs are mainly associated with photoreceptor (rods and cones) dysfunction and loss that eventually leads to blindness. Retinal pigment epithelium (RPE) cells are also associated with IRDs (e.g., RPE65 gene mutations). Cell death may be exacerbated by the accumulation of reactive oxygen species (ROS) and inflammation [
2]. IRDs can be subdivided, depending on the photoreceptor affected, into rod-dominated dystrophies, cone- dominated dystrophies, and generalized dystrophies involving both rods and cones [
3]. IRDs present high genetic and clinical heterogeneity. More than 300 genes with autosomal dominant and recessive, X-linked, or mitochondrial inheritance patterns are linked to this group of diseases [
4]. Moreover, different mutations in one gene can result in different phenotypes. IRDs include syndromic forms such as Usher syndrome and non-syndromic forms such as Retinitis Pigmentosa (RP), Leber’s congenital amaurosis, Stargardt’s macular dystrophy, choroideremia, macular degeneration, or congenital stationary night blindness. This heterogeneity makes difficult to find the specific mutation, as well as the correct treatment for most of these diseases.
RP is the most common IRD, with a prevalence of approximately 1 in 4000 individuals [
5]. Patients with RP typically lose night vision in adolescence, peripheral vision in young adulthood, and central vision later in life. At the cellular level, RP is characterized by a progressive degeneration of rod photoreceptors at early stages due to genetic mutations. Rod degeneration causes the release of inflammatory molecules—free radicals, among others—that cause a hostile microenvironment that eventually affects the cones’ survival [
6].
2. Retinal Inflammation in IRDs
2.1. Ocular Inflammation
Ocular inflammation is a common cause of visual impairment. Inflammation is the immune system’s response to a harmful stimulus that is caused by different factors, mainly pathogens, cell damage, toxic metabolites, and even genetic stressors. It is a defense mechanism to recover tissue homeostasis [
7]. The stimuli can induce acute inflammation (short period) or chronic inflammation (extended period), activating different signaling pathways. During acute inflammatory responses, the damage is minimized or eliminated. However, acute, uncontrolled, and excessive inflammation may become chronic and harmful to cells [
7,
8,
9].
Neuroinflammation is the inflammation of the nervous tissue. It is considered a chronic inflammation with sustained activation of glial cells (microglia, astrocytes) and recruitment of other immune cells into the nervous tissue (brain, spinal cord, or retina) [
10]. Microglia cells, the resident innate immune cells in the retina and other nervous tissue, are potential cellular regulators of inflammation [
11]. They perform immune surveillance and they are activated upon pathologic signals. Under normal conditions or in the absence of damage, microglia cells have a branched morphology with a small round soma, and participate in processes of axonal growth, synaptic remodeling, and neuronal survival. In response to harmful stimuli, tissue damage, or free radicals, microglia cells move to a reactive state, which is characterized by the acquisition of an amoeboid morphology, acquiring the characteristic functions of macrophages such as phagocytosis, induction of the inflammatory process, and presentation of antigens to lymphocytes [
9,
12,
13,
14]. Activated microglia and macrophages secrete pro-inflammatory mediators (cytokines, chemokines, etc.) and upregulate the expression of inducible nitric oxide synthase (iNOS) that act on other cells (e.g., photoreceptor cells) [
15]. The nuclear factor kappa beta (NF-kB), mitogen-activated protein kinase (MAPK), or Janus kinase/signal transducer and activator transcription (JAK-STAT) signaling pathways are the most common pathways activated after a harmful stimulus. These signaling pathways promote the production of different inflammatory mediators such as the pro-inflammatory interleukins (ILs) IL1-β, IL-6, IL-8, and IL-12; tumor necrosis factor alpha (TNFα); gamma interferon (IFN-γ); granulocyte monocyte colony-stimulating factor (GM-CSF); anti-inflammatory cytokines (IL-4, IL-10, IL-11), transforming growth factor beta (TGF-β); and inflammatory proteins such as C-reactive protein (CPR), haptoglobulin, serum amyloid protein, fibrinogen, and alpha-1 acid glycoprotein.
2.2. Inflammation in RP
2.2.1. Local Inflammation
In the retina, the chronic inflammation or neuroinflammation causes neuronal damage and leads to the development or progression of a variety of neurodegenerative diseases including IRDs, retinal neovascular disease [
16], diabetic retinopathy (DR), or age-related macular degeneration (AMD) [
17]. In these degenerative retinal diseases, there is specific damage at the onset of each disease, but studies suggest that chronic and low-grade inflammation is also involved in their progression.
In RP, sustained and chronic inflammation is observed in parallel with the progression of the disease in patients and
rd10 mice, a murine model of autosomal recessive RP [
18,
19]. Evidence of inflammatory cell infiltration, including microglia, macrophages, natural killer cells, and monocytes has been reported in the vitreous humor and in the retina of patients with RP [
20,
21,
22]. Microglia activation and upregulation of inflammatory mediators such as TNFα, IL-6, IL1α, IL1β, monocyte chemoattractant proteins 1 and 2 (MCP-1, MCP-2), or platelet growth factor (PGF) are present in both patients and murine models of RP at early stages of the disease, and they are postulated to contribute to photoreceptor death [
23,
24,
25]. Activated macrophages and glial cells release the pro-inflammatory cytokine TNFα, which participates in the induction and maintenance of the immune response. It may be involved in both cell death and survival pathways (see ) [
26]. Moreover, TNFα is a negative regulator of the transcription factor orthodenticle homeobox 2 (OTX2) expressed in RPE that is critical for the outer retina maintenance [
27]. Upregulation of TNFα is found in several inflammatory eye diseases including RP, glaucoma, uveitis, retinal vascular tumors, Adamantiades–Behcet disease, diabetic macular edema, DR, or AMD [
24,
28,
29,
30,
31,
32,
33,
34,
35,
36]. In particular, elevated TNFα content is present in the aqueous humor of patients and in the retina of murine models of RP [
37,
38,
39,
40]. The blockade of TNFα expression in T17M mice with a rhodopsin (RHO) mutation (a model of RP) or the use of antibodies against TNFα (e.g., adalimumab) in
rd10 mice slow down photoreceptor death [
28,
38,
41], suggesting that it may be an attractive therapeutic target.
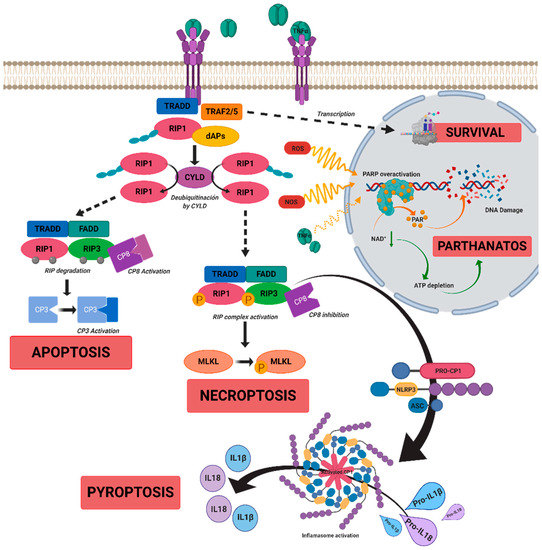
Figure 1. A scheme of possible cell death mechanisms during of tumor necrosis factor alpha (TNFα)-induced signaling in inherited retinal dystrophies (IRDs). TNFα can simultaneously activate multiple signaling pathways of cell death or survival. TNFα binds to tumor necrosis factor receptor 1 (TNFR1), triggering three functional states. (i) The intracellular domain of TNFR1 recruits a death-domain containing adaptor protein (TRADD). TRADD recruits TNF receptor-associated factor 2 (TRAF2) and receptor-interacting protein kinase 1 (RIPK1) to form Complex 1. Complex 1 seems to be important for nuclear factor kappa beta (NF-κB)activation. NF-κB regulates anti-apoptotic genes to block the initiation of apoptosis by Complex 2. (ii) Complex 1 dissociates from TNFR1 and integrates Fas-associated protein with death domain (FADD) and pro-caspase 8 to form Complex 2. The FADD/caspase 8 association depends on complexes containing unubiquitinated RIPK1 as a scaffold. Activated caspase 8 induces caspase 3 and apoptosis. Under apoptotic conditions, active caspase 8 prevents further necroptotic signaling by cleaving and inactivating RIPK1 and RIPK3. (iii) Necroptosis is also mediated through TNFα signaling, when caspase 8 is not active. RIPK1 recruits RIPK3 to form the necrosome complex. RIPK3 phosphorylates the pseudokinase kinase-like domain of mixed-lineage kinase do-main-like (MLKL), leading to its oligomerization. Thus, MLKL recruitment to the plasma membrane induces necroptosis by triggering Ca
+ and Na
2+ influx into the cell. RIPK3 can also promote the nucleotide-binding oligomerization domain containing protein (NOD)-like receptor (NLR) family protein 3 (NLRP3) inflammasome and interleukin (IL)-1β inflammatory responses. TNFα or oxidative stress could activate parthanatos through the overactivation of poly [ADP-ribose] polymerase 1 (PARP1). PARP1 cleaves nicotinamide adenine dinucleotide (NAD+) to nicotinamide and adenosine diphosphate (ADP-ribose). PARPs couple one or more ADP-ribose (PAR) to acceptor proteins (PARylation) and components of the DNA repair machinery. Overactivation of PARP1 can deplete cellular adenosine triphosphate (ATP) and NAD+ storage and lead to bioenergetic collapse and cell death. Adapted from Olivares-González et al. [
28].
2.2.2. Peripheral Inflammation
The eye has an immune privilege to limit the extent of immune responses and to preserve vision [
42]. Ocular fluids and cells have soluble and cell-bound inhibitory factors and immunosuppressive factors including TGF-β2, retinoic acid, galectins, etc. to maintain the immune privilege [
15]. Recent studies show not only local ocular inflammation but also peripheral inflammatory responses in RP patients with high serum IL8, regulated upon activation, normal T cell expressed, and secreted chemokine (C-C motif) ligand 5 (CL5), and CPR levels that correlate with an impairment of the visual function [
18,
25]. Therefore, systemic inflammation could also contribute to the progression of RP [
18]. Moreover, studies in animal models suggest that peripheral damage, such as a systemic infection or a chronic inflammatory process, may also play a role in retinal degeneration in patients with IRDs. Thus, this should be taken into account in the development of possible treatments [
43].
3. Conclusions and Future Perspectives
The inflammatory processes and the putative cell death mechanisms related to inflammation during IRDs, mainly RP, were reviewed. The main drivers of the death of photoreceptor (or RPE) cells in IRDs depend on the genetic defect, the cell type, and the stage of the disease. However, there are other underlying processes that contribute to the progression of retinal degeneration, including inflammation, oxidative stress, dysregulation of cGMP, intracellular calcium accumulation, or ER stress, among others [
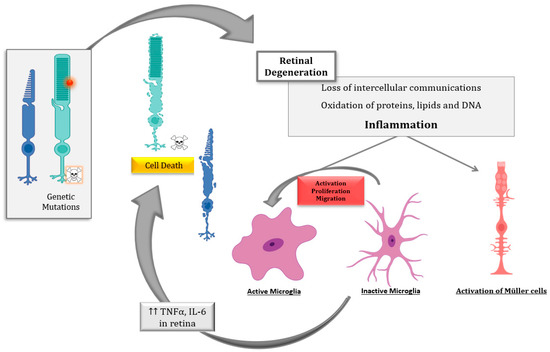
6]. Several pieces of evidence propose that inflammation is critical to both the development and exacerbation of IRDs ().
Figure 2. Schematic representation of the inflammatory processes underlying photoreceptor degeneration in retinitis pigmentosa, the most common inherited retinal dystrophy (IRD). The genetic defect leads to rod degeneration. During rod degeneration several cellular processes are activated, including inflammation and oxidative stress, which lead to the loss of intercellular communications, the oxidation of macromolecules, and the activation of microglia and Müller cells, among others. Activated microglia cells release inflammatory molecules such as cytokines (tumor necrosis factor alpha (TNFα) and interleukin 6 (IL6)), chemokines, etc., which would exacerbate photoreceptor degeneration (both rods and cones) through different cell death mechanism such as necroptosis, parthanatos, apoptosis, etc.
Despite the advances in the knowledge of the cell death mechanisms involved in IRDs, the diversity of the results highlights the great complexity underlying IRDs. Several cell death mechanisms including apoptosis, necroptosis, pyroptosis, and parthanatos, among others, may be involved in photoreceptor degeneration. Inflammation and cell death are tightly interconnected and both influence each other. Therefore, the development of therapies based on ameliorating the inflammatory processes and associated cell mechanisms is crucial for slowing down IRDs. These therapies could be promising mutation-independent strategies.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22042096