Female infertility is mainly caused by ovulation disorders, which affect female reproduction and pregnancy worldwide, with polycystic ovary syndrome (PCOS) being the most prevalent of these. PCOS is a frequent endocrine disease that is associated with abnormal function of the female sex hormone estrogen and estrogen receptors (ERs). Estrogens mediate genomic effects through ERα and ERβ in target tissues. The G-protein-coupled estrogen receptor (GPER) has recently been described as mediating the non-genomic signaling of estrogen. Changes in estrogen receptor signaling pathways affect cellular activities, such as ovulation; cell cycle phase; and cell proliferation, migration, and invasion. Over the years, some selective estrogen receptor modulators (SERMs) have made substantial strides in clinical applications for subfertility with PCOS, such as tamoxifen and clomiphene, however the role of ER in PCOS still needs to be understood.
- estrogen
- estrogen receptor
- ovary
- polycystic ovary syndrome
1. Introduction
Polycystic ovary syndrome (PCOS) is a type of general disease in women that is associated with a variety of reproductive and metabolic disorders [1]. The symptom is characterized by arrested folliculogenesis, hyperandrogenism, and polycystic ovaries [2]. Women suffering from PCOS account for a large proportion of the world population, with the prevalence estimated at between 5% and 10% [3]. Since the National Institutes of Health established a more standardized diagnosis standard in 1990, and with the continuous revision of this standard, the prevalence of PCOS is now expected to be close to 18% in the United States [4]. People with PCOS have more adverse reproductive risks, such as increased incidence of implantation failure, recurrent abortion, spontaneous abortion, premature birth, and endometrial carcinoma [5][6]. For example, women with PCOS were shown to have a 2.7-fold increased risk of endometrial cancer [7]. In addition to reproductive abnormalities, PCOS is also closely related to large quantities of metabolic disorders, for instance increased risk of hyperlipidemia, diabetes mellitus type II (T2DM), and hypertension, as well as hepatic steatosis, glucose intolerance, and insulin resistance [8][9].
Normal ovulation is the result of synergy between follicle-stimulating hormone (FSH) and luteinizing hormone (LH). LH stimulates the theca cells of the ovarian follicle, leading to androgen synthesis. Some of these are bound to sex-hormone-binding globulin (SHBG), and some androgens spread to nearby granulosa cells (GCs), where they are converted to estrogen under the stimulation of FSH. This causes an increase in the level of estrogen hormones, which in turn generates positive feedback in the form of LH production, causing a surge in LH and triggering ovulation [10]. The proliferation and differentiation of theca cells and granulosa cells are regulated by locally produced growth factors. Therefore, ovarian follicular development is tightly regulated by many hormones and other growth factors [11]. After ovulation, the corpus luteum is formed, which secretes the steroid hormones progesterone and estrogen, and then makes the endometrium more receptive to implantation [12].
Research on women with PCOS indicates elevated production of LH and free testosterone. Under the influence of endocrine-disrupting chemicals (phytoestrogens or synthetic estrogenic compounds), hormone secretion is also further affected, resulting in the PCOS disease [13]. As its name implies, the disease is a state of chronic anovulation that involves many ovarian cysts, characterized by an increased number of immature cystic follicles [14]. The ovaries are the primary site of estrogen synthesis, mainly producing 17β-estradiol (E2), which exerts effects on target organs and cells via multiple estrogen receptors (ERs), ERα, ERβ, and the G-protein-coupled estrogen receptor (GPER, also known as GPR30) to maintain various stages of normal development in the human ovaries and uterus.
2. The Characteristics of Estrogen Receptors
2.1. Estrogen Receptors: Expression, Structure
Estrogen mediates most biological effects at the gene level through estrogen receptors; maintains normal reproductive function; and plays key roles in the musculoskeletal, cardiovascular, immune, and central nervous systems [15]. As members of the nuclear receptor family of transcription factors, estrogen receptors occur not only in the nucleus, but also in the cytoplasm and mitochondria of cells. In 1958, Elwood Jensen discovered the first ER (known as ERα). It was subsequently demonstrated that conjugated estrogen receptors could migrate to the nucleus, thereby stimulating gene transcription [16][17]. Similarly, a second ER gene, ERβ, was discovered by Jan-Ake Gustafsson in 1996 [18]. They discovered that ERβ was significantly highly expressed in the prostate and ovaries of rats by in situ hybridization, and that it is highly homologous to ERα. According to the current level of knowledge, the tissue distribution of the ER types is different. ERα is mainly expressed in the uterus, with a small amount expressed in the skin, ovaries, testis, and gut, whereas the expression of ERβ is found in the ovaries, prostate, colon, kidneys, cardiovascular system, and central nervous system (CNS) [19].
ERα and ERβ are different receptors encoded by Esr1 and Esr2 on different chromosomes. ERα and ERβ are respectively found on human chromosome 6 and chromosome 14. The ERα protein is composed of 595 amino acids, with approximate molecular weight of 67 kDa. The full-length size of ERβ is 530 amino acids, with an approximate molecular weight of 59 kDa. Due to selective splicing of transcripts, they may occur in multiple isoforms. There are three ERα isoforms that have been identified. ERαΔ3 lacks exon 3, which encodes part of the DNA-binding domain. ERα36 lacks transcriptional activation domains (AF-1 and AF-2), but retains the DNA-binding domain and partial dimerization and ligand-binding domains [20]. ERα46 is produced by the selective splicing of an ERα gene product, which causes the start codon of exon 2 to initiate protein translation [21]. Four ERβ isoforms, ERβ2, ERβ3, ERβ4, and ERβ5, have been described [22]. All ERβ variants have a new C-terminus and do not bind to estrogen ligands that have been studied (as shown in Figure 1).
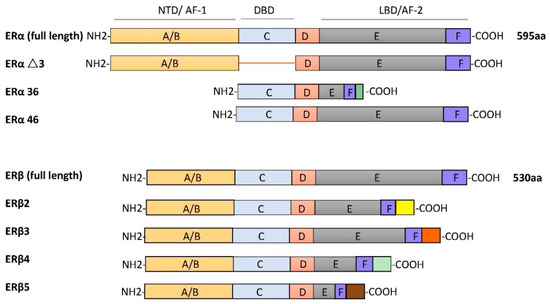
Figure 1. The schematic structures of estrogen receptor (ER) isoforms. Structural domains of ERα and ERβ are labeled A–F. Different functional domains are highlighted as follows: N- terminal (NTD/AF-1) in orange, DNA binding domain (DBD, C domain) in blue, the hinge (D domain) in red, and ligand-dependent transactivation function 2 (LBD/AF-2) in gray/dark blue.
The structures of ERα and ERβ are composed of different functional domains. The main functional domains are called A/B, C, D, and E/F, which also have high sequence homology. The A/B region is the amino terminal domain (NTD), the C region corresponds to the DNA-binding domain (DBD), and the D domain is a hinge region connecting the C and E domains, capable of binding to chaperones. The carboxy terminal region contains the E/F region, also known as the ligand-binding domain (LBD) [23]. In silico analysis shows that two types of ERs have a common structural domain, namely the N terminal, DBD, and the C terminal, LBD [24]. These two receptors belong to the nuclear superfamily and function principally to regulate all kinds of cellular processes, such as proliferation, survival, differentiation, and apoptosis.
Another estrogen receptor, originally identified from human and rat tissues, is named orphan GPR30 [25][26]. Subsequent studies showed that GPR30 can specifically bind to estrogen and is a typical estrogen membrane receptor [27][28]. In 2007, GPR30 was officially named GPER, which has been extensively studied for its role in mediating a rapid response to estrogen, as well as overall physiological and pathological processes in human and animal models [29]. The gene encoding the membrane receptor GPER is located in chromosome 7. As a typical G-protein-coupled receptor, its structure consists of seven trans-membrane α-helical regions, four extracellular segments, and four cytosolic segments [30]. This receptor has a lower estrogen-binding affinity than other estrogen receptors. However, GPER is responsible for the rapid estrogen-mediated activation of ERK1/2 [31]. GPER is widely found in numerous human tissues, such as the reproductive system, prostate, ovaries, and placenta, as well as in the heart, liver, lungs, adipose tissue, and blood vessels [32].
2.2. Estrogen Receptor Ligands
Natural endogenous estrogens are mostly produced in the ovaries, corpus luteum, and placenta, predominantly 17β-estradiol, which is the main ligand of ERs. Various natural and man-made chemicals also have estrogenic activity. Phytoestrogens are plant-derived compounds with structural similarity to 17β-oestradiol, including isoflavones, prenylflavonoids, coumestans, and lignans [33][34]. Synthetic estrogenic compounds (also known as environmental estrogens) include pesticides, dioxins, phthalates, bisphenol A, and diethylstilbestrol [35][36]. They are often widely dispersed in the environment and result in developmental and reproductive abnormalities in humans.
It is believed that most phytoestrogens and synthetic estrogenic compounds exert their physiological effects by regulating ERα and ERβ [37]. Many of these compounds can also activate GPER, including the soy isoflavone genistein, nonylphenol, and bisphenol A [38].
Other compounds are also widely used in clinical and therapeutic applications. For example, selective estrogen receptor modulators (SERMs) are synthetic non-steroidal drugs that act as both ER agonists and ER antagonists [39][40]. Common selective estrogen receptor modulators are tamoxifen, raloxifene, and clomiphene citrate (CC). By contrast, fulvestrant is a selective estrogen receptor downregulator (SERD) that results in ER degradation or downregulation and blocks the proliferation of breast cancer cells [41]. Apart from this, the non-steroidal ligand G-1 (1-[4-(6-bromobenzo[1,3] dioxol-5yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl]-ethanone), which acts as a selective agonist for GPER, has been shown to induce the expression of genes by activation of GPER rather than the classical ERα or ERβ (as shown in Figure 2).
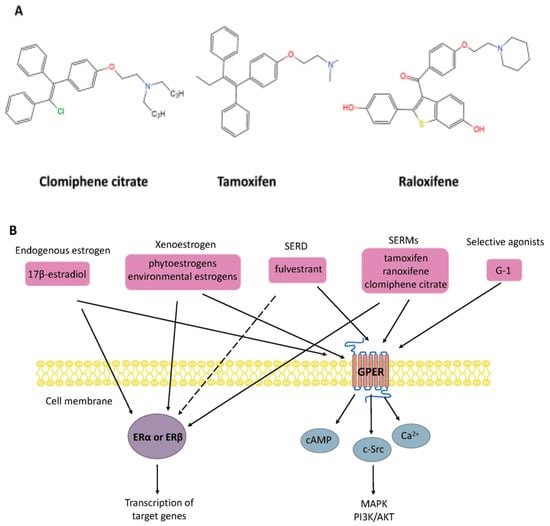
Figure 2. (A) Chemical structure of selective estrogen receptor modulators clomiphene citrate, tamoxifen, and raloxifene. (B) Nongenomic and genomic estrogen signal transduction pathways. Estrogen receptor ligands include 17β-estradiol and other compounds. Dashed lines indicate tissue-specific inhibition. SERD, selective estrogen receptor downregulator; SERM, selective estrogen receptor modulators; G-1, 1-[4-(6-bromobenzo[1,3] dioxol-5yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl]-ethanone; ER, estrogen receptor; GPER, G-protein-coupled estrogen receptor; Ca2+, calcium ions; cAMP, cyclic adenosine monophosphate; MAPK, mitogen-activated protein kinase; PI3K/AKT, phosphatidylinositol 3-kinase/protein kinase B.
3. Functions of ERs in PCOS
3.1. ERs with Follicular Formation/Ovulation
Although it is generally believed that disorders of the hypothalamic–pituitary–ovary axis are a significant cause of cystic formation, the cystic follicle maintains a static condition without degeneration after ovulation failure, which is considered another reason for the development of cysts [42]. In mice, abnormal ERβ expression in the ovaries results in the failure of dominant follicles to develop consistently [43]. Ovarian estrogens are thought to regulate follicular maturation locally in the ovaries and to stimulate the proliferation of GC during dominant follicle growth. Recently, the relationship between GPER and oocyte maturation was reported. The research study showed that the maturation of carp oocytes significantly decreased when they were incubated with either E2 or GPER agonist G-1 [44]. Another study showed that in cumulus granulosa cells from patients with PCOS, low levels of E2, accompanied by high levels of GPER, might inhibit human oocyte maturation [45]. Meanwhile, GPER small interfering RNA knockdown mice indicated that GPER had a stronger inhibitory action on primordial follicles, indicating that GPERs might play a crucial part in oocyte maturation [46]. Estrogens inhibit meiotic maturation of full-grown oocytes by activating the estrogen receptor GPER.
Estrogen has been reported to affect some ovarian functions via autocrine or paracrine actions, most significantly enhancing the effect of FSH on granulosa cells [47]. ERs are expressed in GC and theca cells (TC) in developing follicles. Based on these considerations, in conditions characterized by ovulation dysfunction, such as PCOS, altered ovarian ER expression may have an essential role. Mice with the ERα gene disrupted developed a phenotype similar to PCOS, with high circulating LH concentrations and ovaries characterized by multiple hemorrhagic and cystic follicles with non-ovulation [48][49]. Likewise, by detecting mRNA and protein levels of ERα and ERβ in GC and TC from regularly circulating ovaries and polycystic human ovaries, we observed that the expression of ERα in GC was higher than in TC, but ERβ was expressed similarly in both cell types [50]. It seems that ERα has a significant advantage compared to ERβ in poor follicular development and ovulation failure in ovary syndrome. Rumi et al. used zinc finger nuclease (ZFN)-mediated genome editing to generate ER knockout rats, in which they could observe the phenomena of uterine dysplasia, polycystic ovaries, and ovulation defects [51]. Specifically, these animals failed to express functional ERα proteins and showed obvious abnormalities in postnatal growth, fertility, female genital tract development, and response to E2. In humans, an 18-year-old woman was reported to have a homozygous ESR1 mutation in a completely conserved residue, with an ovarian cyst and a small uterus despite having elevated circulating serum E2 [52]. Interestingly, ERβ knockout mice also showed morphological characteristics of abnormal follicular development, as well as decreased ovulation ability [53]. Compared with wild-type mice, they had earlier atretic follicles and fewer corpora lutea, although ER gene knockout mice can reproduce [54][55]. Similarly, another experiment also showed female ERβ knockout mice to have poor fertility, characterized by lower ovulation numbers, fewer pregnancies, and preovulatory follicles exhibiting a weak response to FSH-induced differentiation [56]. Thus, abnormal expression of ERs (ERα and ERβ) may be related to poor oocyte development and ovulatory failure in PCOS patients.
3.2. ER Changes Associated with Endometrium
During normal menstrual cycles, the endometrium undergoes rapid cycling, proliferation, and growth in response to estrogen through the action of specific steroid receptors ERα and the ERβ [57]. During the follicular phase, E2 induces the rapid growth of the uterine endometrium and increases endometrial sensitivity to estrogen by increasing ERα levels. Conversely, at the luteal phase of the menstrual cycle, the corpus luteum continuously secretes progesterone and reduces estrogen to provide an appropriate uterine environment for maintaining the pregnancy. Thus, improper estrogen action affecting maximal uterine acceptance capability may change the normal expression of genes and reduce fertility in women with PCOS or increase the rate of spontaneous miscarriage.
ERα and the ERβ have different cellular localizations in the human endometrium. In general, ER (ERα and ERβ) expression reaches a maximum at the late proliferative stage and decreases at the secretory phase of the menstrual cycle [58][59]. For example, ERα mRNA is expressed in both the endometrial epithelial and stromal cells during the menstrual cycle, whereas ERβ mRNA is found predominantly in glandular epithelial cells [60]. In addition, the expression level of ERα mRNA in the uterus is more prominent than ERβ mRNA. GPER is localized in the plasma membrane and in the endoplasmic reticulum, which is considered to regulate the growth and proliferation of endometrial cells through its interaction with ERα [61][62][63].
There is growing clinical and experimental evidence showing that ER is an endometrial marker in patients with PCOS. Estrogen-induced uterine hyperplasia occurs by binding uterine epithelial ERα in adult mice to actively inhibit epithelial apoptosis in the uterus [64]. The same scenario is observed for ovulatory PCOS, where high levels of ERα expression are observed. In women with PCOS, ERα expression in the endometrium continues to enter the secretion stage, showing high expression in the stroma and luminal epithelium [65]. One woman with a homozygous ERα mutation and rats lacking ERα showed similar phenotypes and infertility similar to what has been observed in patients with PCOS [49][52].
In women with ovulatory dysfunction, the endometrium, as a target tissue for estrogen, is prone to hyperplasia and cancer [66][67][68]. PCOS symptoms are significantly related to endometrial cancer risk. Women under age 50 with PCOS have four times higher incidence of endometrial cancer than women without PCOS [69]. Based on previous research, increased ERα expression in the endometrium of PCOS women at both gene and protein expression levels has been observed, suggesting that the endometrium is more sensitive to estrogen, possibly explaining the significant increased incidence of hyperplasia and endometrial cancer, as well as the decreased ability to continue pregnancy [70][71]. Moreover, an ERβ polymorphism (+1730 G/A) was associated with the development of susceptibility to PCOS in humans [72]. Polymorphisms in identified ER-α were found to have an association with the development of endometrial cancer [73]. Furthermore, changes in ERα expression levels and the ERα/ERβ ratio in PCOS patients were higher than in the normal group, indicating outstanding ERα-mediated actions in the endometrium, which may be related to endometrial hyperplasia and endometrial cancer [74]. Previous studies have shown that p160 steroid receptor coactivator is increased in the secretory-phase endometria of women with PCOS, which further increases endometrial ERα expression to stimulate endometrial proliferation [74][75]. Abnormal estrogenic environments may alter endometrial receptivity in women, resulting in blastocyst implantation failure or a malformed abortion after implantation (the functions of ERs in PCOS are shown in Figure 3).
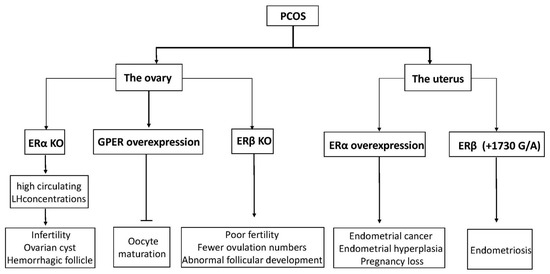
Figure 3. A diagram of physiological phenomena to explain disordered expression in the ER family. Arrows indicate direct effects, while no arrows indicate inhibition.
This entry is adapted from the peer-reviewed paper 10.3390/cells10020459
References
- Rosenfield, R.L.; Ehrmann, D.A. The Pathogenesis of Polycystic Ovary Syndrome (PCOS): The Hypothesis of PCOS as Functional Ovarian Hyperandrogenism Revisited. Endocr. Rev. 2016, 37, 467–520.
- Dumesic, D.A.; Oberfield, S.E.; Stener-Victorin, E.; Marshall, J.C.; Laven, J.S.; Legro, R.S. Scientific Statement on the Diagnostic Criteria, Epidemiology, Pathophysiology, and Molecular Genetics of Polycystic Ovary Syndrome. Endocr. Rev. 2015, 36, 487–525.
- Khan, M.J.; Ullah, A.; Basit, S. Genetic Basis of Polycystic Ovary Syndrome (PCOS): Current Perspectives. Appl. Clin. Genet. 2019, 12, 249–260.
- March, W.A.; Moore, V.M.; Willson, K.J.; Phillips, D.I.W.; Norman, R.J.; Davies, M.J. The prevalence of polycystic ovary syndrome in a community sample assessed under contrasting diagnostic criteria. Hum. Reprod. 2010, 25, 544–551.
- Azziz, R.; Carmina, E.; Chen, Z.; Dunaif, A.; Laven, J.S.E.; Legro, R.S.; Lizneva, D.; Natterson-Horowtiz, B.; Teede, H.J.; Yildiz, B.O. Polycystic ovary syndrome. Nat. Rev. Dis. Primers. 2016, 2, 16057.
- Boomsma, C.M.; Eijkemans, M.J.C.; Hughes, E.G.; Visser, G.H.A.; Fauser, B.C.J.M.; Macklon, N.S. A meta-analysis of pregnancy outcomes in women with polycystic ovary syndrome. Hum. Reprod. Update 2006, 12, 673–683.
- Dumesic, D.A.; Lobo, R.A. Cancer risk and PCOS. Steroids 2013, 78, 782–785.
- Cobin, R.H. Cardiovascular and metabolic risks associated with PCOS. Intern. Emerg. Med. 2013, 8 (Suppl. 1), S61–S64.
- Azziz, R. Polycystic Ovary Syndrome. Obstet. Gynecol. 2018, 132, 321–336.
- Richards, J.S.; Pangas, S.A. The ovary: Basic biology and clinical implications. J. Clin. Investig. 2010, 120, 963–972.
- Liu, Y.; Zhang, Y.; Li, Y.; Liu, X.; Wang, X.; Zhang, C.; Hao, C.; Deng, S. Regulation of follicular development and differentiation by intra-ovarian factors and endocrine hormones. Front. Biosci. (Landmark Ed.) 2019, 24, 983–993.
- Kumar, P.; Sait, S.F. Luteinizing hormone and its dilemma in ovulation induction. J. Hum. Reprod. Sci. 2011, 4, 2–7.
- Tang, Z.; Xu, X.; Deng, S.; Lian, Z.; Yu, K. Oestrogenic Endocrine Disruptors in the Placenta and the Fetus. Int. J. Mol. Sci. 2020, 21, 1519.
- Li, Y.; Guo, J.; Deng, S.; Gao, Z.; Liu, Y.; Gu, Q. Fibrin Facilitates Mesenchymal Stem Cells to Ameliorate Rats with Polycystic Ovary Syndrome. Appl. Sci. 2020, 10, 3598.
- Gustafsson, J. What pharmacologists can learn from recent advances in estrogen signalling. Trends Pharmacol. Sci. 2003, 24, 479–485.
- Jensen, E.V.; Desombre, E.R.; Kawashima, T.; Suzuki, T.; Kyser, K.; Jungblut, P.W. Estrogen-binding substances of target tissues. Science 1967, 158, 529–530.
- Jensen, E.V.; Suzuki, T.; Kawashima, T.; Stumpf, W.E.; Jungblut, P.W.; DeSombre, E.R. A two-step mechanism for the interaction of estradiol with rat uterus. Proc. Natl. Acad. Sci. USA 1968, 59, 632–638.
- Kuiper, G.G.; Enmark, E.; Pelto-Huikko, M.; Nilsson, S.; Gustafsson, J.A. Cloning of a novel receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. USA 1996, 93, 5925–5930.
- Brandenberger, A.W.; Tee, M.K.; Lee, J.Y.; Chao, V.; Jaffe, R.B. Tissue distribution of estrogen receptors alpha (ER-alpha) and beta (ER-beta) mRNA in the midgestational human fetus. J. Clin. Endocrinol. Metab. 1997, 82, 3509–3512.
- Shi, L.; Dong, B.; Li, Z.; Lu, Y.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B.; et al. Expression of ER-{alpha}36, a novel variant of estrogen receptor {alpha}, and resistance to tamoxifen treatment in breast cancer. J. Clin. Oncol. 2009, 27, 3423–3429.
- Denger, S.; Reid, G.; Kos, M.; Flouriot, G.; Parsch, D.; Brand, H.; Korach, K.S.; Sonntag-Buck, V.; Gannon, F. ERalpha gene expression in human primary osteoblasts: Evidence for the expression of two receptor proteins. Mol. Endocrinol. 2001, 15, 2064–2077.
- Moore, J.T.; McKee, D.D.; Slentz-Kesler, K.; Moore, L.B.; Jones, S.A.; Horne, E.L.; Su, J.L.; Kliewer, S.A.; Lehmann, J.M.; Willson, T.M. Cloning and characterization of human estrogen receptor beta isoforms. Biochem. Biophys. Res. Commun. 1998, 247, 75–78.
- Jia, M.; Dahlman-Wright, K.; Gustafsson, J. Estrogen receptor alpha and beta in health and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 557–568.
- Kumar, R.; Zakharov, M.N.; Khan, S.H.; Miki, R.; Jang, H.; Toraldo, G.; Singh, R.; Bhasin, S.; Jasuja, R. The dynamic structure of the estrogen receptor. J. Amino Acids 2011, 2011, 812540.
- Feng, Y.; Gregor, P. Cloning of a novel member of the G protein-coupled receptor family related to peptide receptors. Biochem. Biophys. Res. Commun. 1997, 231, 651–654.
- O’Dowd, B.F.; Nguyen, T.; Marchese, A.; Cheng, R.; Lynch, K.R.; Heng, H.H.; Kolakowski, L.F.J.; George, S.R. Discovery of three novel G-protein-coupled receptor genes. Genomics 1998, 47, 310–313.
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005, 307, 1625–1630.
- Thomas, P.; Pang, Y.; Filardo, E.J.; Dong, J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 2005, 146, 624–632.
- Prossnitz, E.R.; Barton, M. Estrogen biology: New insights into GPER function and clinical opportunities. Mol. Cell Endocrinol. 2014, 389, 71–83.
- Barton, M.; Filardo, E.J.; Lolait, S.J.; Thomas, P.; Maggiolini, M.; Prossnitz, E.R. Twenty years of the G protein-coupled estrogen receptor GPER: Historical and personal perspectives. J. Steroid Biochem. Mol. Biol. 2018, 176, 4–15.
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R.J. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660.
- Qian, H.; Xuan, J.; Liu, Y.; Shi, G. Function of G-Protein-Coupled Estrogen Receptor-1 in Reproductive System Tumors. J. Immunol. Res. 2016, 2016, 7128702.
- Ososki, A.L.; Kennelly, E.J. Phytoestrogens: A review of the present state of research. Phytother. Res. 2003, 17, 845–869.
- Patisaul, H.B.; Jefferson, W. The pros and cons of phytoestrogens. Front. Neuroendocrinol. 2010, 31, 400–419.
- Lóránd, T.; Vigh, E.; Garai, J. Hormonal action of plant derived and anthropogenic non-steroidal estrogenic compounds: Phytoestrogens and xenoestrogens. Curr. Med. Chem. 2010, 17, 3542–3574.
- Casals-Casas, C.; Desvergne, B. Endocrine disruptors: From endocrine to metabolic disruption. Annu. Rev. Physiol. 2011, 73, 135–162.
- Singleton, D.W.; Khan, S.A. Xenoestrogen exposure and mechanisms of endocrine disruption. Front. Biosci. 2003, 8, s110–s118.
- Thomas, P.; Dong, J. Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: A potential novel mechanism of endocrine disruption. J. Steroid Biochem. Mol. Biol. 2006, 102, 175–179.
- Jordan, V.C. SERMs: Meeting the promise of multifunctional medicines. J. Natl. Cancer Inst. 2007, 99, 350–356.
- Morello, K.C.; Wurz, G.T.; DeGregorio, M.W. SERMs: Current status and future trends. Crit. Rev. Oncol. Hematol. 2002, 43, 63–76.
- Huang, D.; Yang, F.; Wang, Y.; Guan, X. Mechanisms of resistance to selective estrogen receptor down-regulator in metastatic breast cancer. Biochim. Biophys. Acta. Rev. Cancer 2017, 1868, 148–156.
- Isobe, N.; Yoshimura, Y. Deficient proliferation and apoptosis in the granulosa and theca interna cells of the bovine cystic follicle. J. Reprod. Dev. 2007, 53, 1119–1124.
- Tang, Z.; Zhang, R.; Lian, Z.; Deng, S.; Yu, K. Estrogen-Receptor Expression and Function in Female Reproductive Disease. Cells 2019, 8, 1123.
- Majumder, S.; Das, S.; Moulik, S.R.; Mallick, B.; Pal, P.; Mukherjee, D. G-protein coupled estrogen receptor (GPER) inhibits final oocyte maturation in common carp, Cyprinus carpio. Gen. Comp. Endocrinol. 2015, 211, 28–38.
- Zang, L.; Zhang, Q.; Zhou, Y.; Zhao, Y.; Lu, L.; Jiang, Z.; Peng, Z.; Zou, S. Expression pattern of G protein-coupled estrogen receptor 1 (GPER) in human cumulus granulosa cells (CGCs) of patients with PCOS. Syst. Biol. Reprod. Med. 2016, 62, 184–191.
- Wang, C.; Prossnitz, E.R.; Roy, S.K. G protein-coupled receptor 30 expression is required for estrogen stimulation of primordial follicle formation in the hamster ovary. Endocrinology 2008, 149, 4452–4461.
- Richards, J.S. Hormonal control of gene expression in the ovary. Endocr. Rev. 1994, 15, 725–751.
- Schomberg, D.W.; Couse, J.F.; Mukherjee, A.; Lubahn, D.B.; Sar, M.; Mayo, K.E.; Korach, K.S. Targeted disruption of the estrogen receptor-alpha gene in female mice: Characterization of ovarian responses and phenotype in the adult. Endocrinology 1999, 140, 2733–2744.
- Hamilton, K.J.; Arao, Y.; Korach, K.S. Estrogen hormone physiology: Reproductive findings from estrogen receptor mutant mice. Reprod. Biol. 2014, 14, 3–8.
- Jakimiuk, A.J.; Weitsman, S.R.; Yen, H.; Bogusiewicz, M.; Magoffin, D.A. Estrogen receptor alpha and beta expression in theca and granulosa cells from women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2002, 87, 5532–5538.
- Rumi, M.A.K.; Dhakal, P.; Kubota, K.; Chakraborty, D.; Lei, T.; Larson, M.A.; Wolfe, M.W.; Roby, K.F.; Vivian, J.L.; Soares, M.J. Generation of Esr1-knockout rats using zinc finger nuclease-mediated genome editing. Endocrinology 2014, 155, 1991–1999.
- Quaynor, S.D.; Stradtman, E.W.J.; Kim, H.; Shen, Y.; Chorich, L.P.; Schreihofer, D.A.; Layman, L.C. Delayed puberty and estrogen resistance in a woman with estrogen receptor α variant. N. Engl. J. Med. 2013, 369, 164–171.
- Hewitt, S.C.; Winuthayanon, W.; Korach, K.S. What’s new in estrogen receptor action in the female reproductive tract. J. Mol. Endocrinol. 2016, 56, R55–R71.
- Krege, J.H.; Hodgin, J.B.; Couse, J.F.; Enmark, E.; Warner, M.; Mahler, J.F.; Sar, M.; Korach, K.S.; Gustafsson, J.A.; Smithies, O. Generation and reproductive phenotypes of mice lacking estrogen receptor beta. Proc. Natl. Acad. Sci. USA 1998, 95, 15677–15682.
- Harris, H.A. Estrogen receptor-beta: Recent lessons from in vivo studies. Mol. Endocrinol. 2007, 21, 1–13.
- Couse, J.F.; Yates, M.M.; Deroo, B.J.; Korach, K.S. Estrogen receptor-beta is critical to granulosa cell differentiation and the ovulatory response to gonadotropins. Endocrinology 2005, 146, 3247–3262.
- Shiozawa, T.; Li, S.F.; Nakayama, K.; Nikaido, T.; Fujii, S. Relationship between the expression of cyclins/cyclin-dependent kinases and sex-steroid receptors/Ki67 in normal human endometrial glands and stroma during the menstrual cycle. Mol. Hum. Reprod. 1996, 2, 745–752.
- Punyadeera, C.; Verbost, P.; Groothuis, P. Oestrogen and progestin responses in human endometrium. J. Steroid Biochem. Mol. Biol. 2003, 84, 393–410.
- Snijders, M.P.; de Goeij, A.F.; Debets-Te Baerts, M.J.; Rousch, M.J.; Koudstaal, J.; Bosman, F.T. Immunocytochemical analysis of oestrogen receptors and progesterone receptors in the human uterus throughout the menstrual cycle and after the menopause. J. Reprod. Fertil. 1992, 94, 363–371.
- Matsuzaki, S.; Fukaya, T.; Suzuki, T.; Murakami, T.; Sasano, H.; Yajima, A. Oestrogen receptor alpha and beta mRNA expression in human endometrium throughout the menstrual cycle. Mol. Hum. Reprod. 1999, 5, 559–564.
- Otto, C.; Rohde-Schulz, B.; Schwarz, G.; Fuchs, I.; Klewer, M.; Brittain, D.; Langer, G.; Bader, B.; Prelle, K.; Nubbemeyer, R.; et al. G protein-coupled receptor 30 localizes to the endoplasmic reticulum and is not activated by estradiol. Endocrinology 2008, 149, 4846–4856.
- Funakoshi, T.; Yanai, A.; Shinoda, K.; Kawano, M.M.; Mizukami, Y. G protein-coupled receptor 30 is an estrogen receptor in the plasma membrane. Biochem. Biophys. Res. Commun. 2006, 346, 904–910.
- Prossnitz, E.R.; Barton, M. The G-protein-coupled estrogen receptor GPER in health and disease. Nature reviews. Endocrinology 2011, 7, 715–726.
- Winuthayanon, W.; Hewitt, S.C.; Orvis, G.D.; Behringer, R.R.; Korach, K.S. Uterine epithelial estrogen receptor α is dispensable for proliferation but essential for complete biological and biochemical responses. Proc. Natl. Acad. Sci. USA 2010, 107, 19272–19277.
- Margarit, L.; Taylor, A.; Roberts, M.H.; Hopkins, L.; Davies, C.; Brenton, A.G.; Conlan, R.S.; Bunkheila, A.; Joels, L.; White, J.O.; et al. MUC1 as a discriminator between endometrium from fertile and infertile patients with PCOS and endometriosis. J. Clin. Endocrinol. Metab. 2010, 95, 5320–5329.
- Pillay, O.C.; Te Fong, L.F.W.; Crow, J.C.; Benjamin, E.; Mould, T.; Atiomo, W.; Menon, P.A.; Leonard, A.J.; Hardiman, P. The association between polycystic ovaries and endometrial cancer. Hum. Reprod. 2006, 21, 924–929.
- Haoula, Z.; Salman, M.; Atiomo, W. Evaluating the association between endometrial cancer and polycystic ovary syndrome. Hum. Reprod. 2012, 27, 1327–1331.
- Villavicencio, A.; Bacallao, K.; Gabler, F.; Fuentes, A.; Albornoz, J.; Casals, A.; Vega, M. Deregulation of tissue homeostasis in endometria from patients with polycystic ovarian syndrome with and without endometrial hyperplasia. Gynecol. Oncol. 2007, 104, 290–295.
- Fearnley, E.J.; Marquart, L.; Spurdle, A.B.; Weinstein, P.; Webb, P.M. Polycystic ovary syndrome increases the risk of endometrial cancer in women aged less than 50 years: An Australian case-control study. Cancer Causes Control 2010, 21, 2303–2308.
- Maliqueo, M.; Clementi, M.; Gabler, F.; Johnson, M.C.; Palomino, A.; Sir-Petermann, T.; Vega, M. Expression of steroid receptors and proteins related to apoptosis in endometria of women with polycystic ovary syndrome. Fertil. Steril. 2003, 80 (Suppl. 2), 812–819.
- Navaratnarajah, R.; Pillay, O.C.; Hardiman, P. Polycystic ovary syndrome and endometrial cancer. Semin. Reprod. Med. 2008, 26, 62–71.
- Kim, J.J.; Choi, Y.M.; Choung, S.H.; Yoon, S.H.; Lee, G.H.; Moon, S.Y. Estrogen receptor beta gene +1730 G/A polymorphism in women with polycystic ovary syndrome. Fertil. Steril. 2010, 93, 1942–1947.
- Kang, S.; Roh, J.W.; Kim, J.W. Single nucleotide polymorphism: A new risk factor for endometrial cancer? Future Oncol. 2005, 1, 323–330.
- Quezada, S.; Avellaira, C.; Johnson, M.C.; Gabler, F.; Fuentes, A.; Vega, M. Evaluation of steroid receptors, coregulators, and molecules associated with uterine receptivity in secretory endometria from untreated women with polycystic ovary syndrome. Fertil. Steril. 2006, 85, 1017–1026.
- Gregory, C.W.; Wilson, E.M.; Apparao, K.B.C.; Lininger, R.A.; Meyer, W.R.; Kowalik, A.; Fritz, M.A.; Lessey, B.A. Steroid receptor coactivator expression throughout the menstrual cycle in normal and abnormal endometrium. J. Clin. Endocrinol. Metab. 2002, 87, 2960–2966.