Post-translational modification with Ubiquitin-like proteins represents a complex signaling language regulating virtually every cellular process. Among these post-translational modifiers is Ubiquitin-fold modifier (UFM1), which is covalently attached to its substrates through the orchestrated action of a dedicated enzymatic cascade. Originally identified to be involved embryonic development, its biological function remains enigmatic. Recent research reveals that UFM1 regulates a variety of cellular events ranging from DNA repair to autophagy and ER stress response implicating its involvement in a variety of diseases. Given the contribution of UFM1 to numerous pathologies, the enzymes of the UFM1 cascade represent attractive targets for pharmacological inhibition.
- UFM1
- Ubiquitin-like modifiers
- substrates
- activity-based probes
1. The UFM1 (de)Conjugation System
Covalent attachment of Ubiquitin or Ubiquitin-like modifiers to their designated substrate proteins, is orchestrated by the sequential action of three enzyme classes: E1, E2, and E3 enzyme. At the apex of this enzymatic cascade, the E1 enzyme activates the C-terminal glycine residue through adenylation and subsequent thioester formation with its active site cysteine residue, poising it for transfer to the active site cysteine of the E2 enzyme. There the Ub or Ubl is activated again and relayed with the cooperation of the E3 ligase to the lysine residue of the substrate [1]. Additionally, self-modification of Ubiquitin and SUMO through one of their lysines can occur forming polymeric chains that further regulate the biological consequence of substrate modification, which is counter-balanced by the action of specific proteases (i.e., de-ubiquitinating enzymes (DUBs), Sentrin-specific proteases (SENPs), de-Neddylases, or UFM1 specific proteases (UFSPs). While the UFM1 activating and conjugating enzymes share the same concept of ATP-dependent activation and trans-thiolation, their architecture differs greatly from the enzymes of the Ubiquitin and Ubiquitin-like modifiers.
1.1. UBA5
Activation of UFM1 through thioester formation by the action of the E1 enzyme (UBA5) at the apex of the enzymatic cascade is key to covalent attachment of this Ubl to its substrates (Figure 1). Firstly, activation of the C-terminal glycine residue occurs through adenylation that then allows the trans-thioesterification through nucleophilic attack of the cysteine residue of the E2 (UFC1) enzyme (Figure 1). Although UBA5, a non-canonical E1 enzyme, lacks a defined active cysteine domain, it contains an adenylation domain harboring the catalytic cysteine [2], reminiscent of Atg7—the E1 enzyme for autophagy-related Ubl LC3—and forms a homodimer through its adenylation domain. Interestingly, UFM1 interaction with the N-terminal UFM1-interacting sequence (UIS) or UFM1-interacting motif (UFIM) adjacent to the adenylation domain of UBA5, is prerequisite to UFM1 activation and subsequent ATP binding as it stabilizes its dimeric state [3][4][5][6] (Figure 1, inset).
Similar to the Ubiquitin-activating enzyme UBE1, human UBA5 has two isoforms differing only by a 56 amino acid long N-terminal extension adjacent to the adenylation domain [6]. Although the short isoform is capable of activating UFM1, biochemical and structural studies have revealed that the N-terminal extension contributes to ATP-binding and modulates the interaction of adenylation domain with ATP such that ATP-gamma phosphate adopts a different structural position compared to the long isoform [5][7][8][9]. In addition to the conformational changes resulting from the lack of the N-terminal region, the catalytic cysteine residue is repositioned on the cross-over loop in the short isoform [6]. Unexpectedly, the presence of the N-terminal region in the long UBA5 isoform increases the ATP binding affinity, resulting in a 1:2 ratio of ATP to UBA5, while the short isoform requires equimolar ATP concentrations [3][5][9]. Prior to relaying UFM1 to the E2 enzyme UFC1, the short C-terminal sequence (UFC1-binding region or UFD domain) of UBA5 recruits the UFM1-thioester intermediate facilitating its adenylation which stimulates trans-thiolation [8] (Figure 1). Moreover, the C-terminal region of UBA5 includes a unique bi-specific LIR/UFIM motif that recognizes both GABARAP and UFM1 through numerous hydrophobic interactions [4] (Figure 1, inset and Figure 2B). Unexpectedly, GABARAP exhibits a ten-fold higher affinity for the LIR/UFIM domain compared to UFM1 and Habisov et al. have reported that its interaction is prerequisite for UBA5 recruitment to the ER membrane [4]. However, GABARAP-mediated translocation of UBA5 to the ER membrane is independent of the lipidation status of this autophagy-related modifier [4][10]. Given that recent research has revealed that the primary UFM1 substrates as well as the E3 ligase UFL1, its adaptor protein DDRGK1 (UFBP1) and the deUFMylase UFSP2 are associated with or anchored in the ER, it is logical that the activating enzyme at the apex of the UFMylation cascade is in proximity. This observation, however, begs the question of how the cytoplasmic E2 enzyme UFC1 translocates to the ER membrane to participate in relaying activated UFM1 to the E3 ligase and ultimately the acceptor lysines of its substrates.
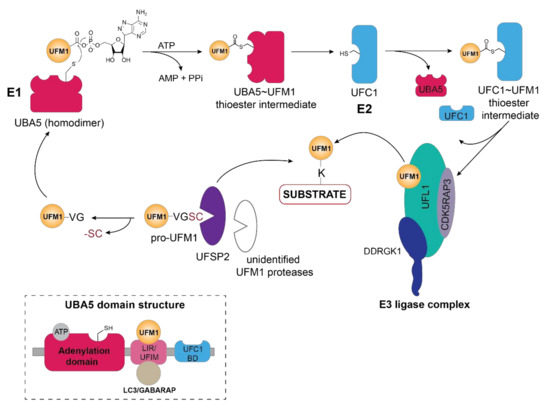
Figure 1. Schematic overview of the UFM1 cascade. In the initial step, the two terminal amino acids (serine and cysteine) are cleaved from pro-UFM1 by UFSP2 and presumably another unidentified UFM1-specific protease [11]. Prior to UFM1 adenylation at the exposed C-terminal glycine residue, UFM1 binds to the UFM1-interacting motif (UFIM)domain of UBA5 (E1), promoting its homodimer formation as well as subsequent UFM1 activation [3]. The UFM1 adenylate intermediate undergoes a nucleophilic attack by the active site cysteine residue of UBA5 yielding the UBA5 ~UFM1 thioester intermediate. Through a transthiolation reaction, activated UFM1 is transferred to the active site cysteine of UFC1 (E2) with the help of the UFC1 binding domain (UFC-BD) of UBA5 [3][4], which then relays the activated UFM1 from the UFC1~UFM1 thioester intermediate via the UFM1 E3 ligase complex to the lysine residue of its substrate proteins. The E3 ligase complex consists of UFL1 and its adaptor proteins DDRGK1 and CDK5RAP3, which stimulate its ligase activity and confer the ER membrane localization of UFL1. Modulation of substrate UFMylation and the resulting cellular response occurs through the action of the UFM1-specific protease UFSP2. Inset depicts the domain organization of UBA5, consisting of the adenylation domain which harbors both the ATP binding pocket and the catalytic site, as well as the LC3-interacting region (LIR) and the UFM1-binding motif (UFIM) and the UFC1-binding domain (UFC1-BD).
1.2. UFC1
Similar to UBA5, the E2-like enzyme UFC1 does not display any conservation with Ubiquitin E2 enzymes and consists of a minimalistic catalytic core domain of 10 amino acids, and an additional N-terminal helix [12][7][13]. Akin to the Ubiquitin E2 enzymes, the active-site cysteine residue catalyzes the trans-thiolation of UFM1 during its transfer from UBA5 (Figure 1). However, structural studies have revealed that the N-terminal helix in UFC1 can adopt a variety of conformations most likely to accommodate different substrates thus conferring substrate specificity [14][15][16]. With predominantly nuclear and partially cytoplasmic localization of the E2-like enzyme UFC1 [12], it is intriguing of whether and how UFC1 localizes to the ER membrane, given that the principal UFMylation substrates and enzymes reside in vicinity of the ER membrane [17].
1.3. UFL1
Akin to ubiquitination, the final step of UFM1 activation and transfer onto the lysine residue of the substrate is orchestrated by the E3 enzyme UFL1, also known as RCAD, Maxer, NLBP, and KIAA0776 (Figure 1). However, UFL1 does not display any homology to the HECT or RING domains of the known Ubiquitin ligases, but rather contains a highly conserved N-terminal domain that is instrumental in UFM1 transfer to the substrate [18][19]. Initially discovered by Tatsumi et al., as an interactor of its one of its substrates, UFL1, which is highly conserved throughout the kingdoms, has been demonstrated to catalyze the UFMylation of numerous substrates, most notably UFBP1 (also referred to as DDRGK1, Dashurin, or C20orf116) [19][20]. Later, it was reported that the UFM1 modification of other substrates such as activating signal co-integrator 1 (ASC1) as well as the ribosomal protein RPL26 is mediated by UFL1 [21][22][23] (Figure 2A). The UFM1 ligase UFL1 consists of a transmembrane-like domain flanked by a nuclear localization signal and a primarily through its C-terminal Proteasome, COP9, Initiation factor 3 (PCI) domain, shared by numerous Proteasome subunits, Cop9 subunits, eIF3 translation initiation factor subunits [20][24]. UFL1 does not display any homology to the domains of the Ubiquitin ligases and lacks a catalytic cysteine in its N-terminal domain commonly found in HECT and RBR class of E3 ligases, it seems to function in a scaffolding manner reminiscent of RING E3 ligases recruiting both the E2 enzyme and the substrate [21]. Recruitment of UFL1 to its main venue—the ER membrane—occurs by virtue of DDRGK1 recruitment through its ER signal peptide—as UFL1 lacks a classical transmembrane domain. Concurrently, interaction between UFL1 and DDRGK1 activates its ligase activity, while bringing it in vicinity of its substrates [20][22][25] (Figure 2A). Deletion of DDRGK1 and subsequent reintroduction of variants lacking the transmembrane domain (TM) underscore its role in promoting the proper subcellular localization of UFL1 [20][22][25]. Intriguingly, a substantial pool of UFL1 resides at the ER membrane where it UFMylates several substrates critical in the UPR (unfolded protein response) pathway, however, it is elusive how the nuclear localization signal sequence (NLS) preceding the PCI domain is exposed to translocate the ligase into the nucleus for UFMylation of targets restricted to the nucleus such as ASC1, MRE11, and Histone H4 [21][26][27] (Figure 2C).
Reminiscent of the Nedd8 modification of the Cullin-RING E3 ligases to activate substrate ubiquitination [28], the UFM1 ligase activity of UFL1 requires interaction with its adaptor protein DDRGK1 (also known as UFBP1, Dashurin, or C20orf116). Directed to the ER membrane through its hydrophobic N-terminal signal sequence and anchored by the adjacent transmembrane (TM) domain, the interaction of DDRGK1 with UFL1 as well as the recruitment of the UFM1-specific protease UFSP2 is mediated by the PCI-domain in a UFM1-dependent manner [19][20]. Although originally identified as an interaction partner of UFL1 [20], the highly conserved protein DDRGK1, has also been observed to be modified with UFM1 within the PCI domain at lysine 267 [19][20], which modulates its binding affinity to UFL1 in turn stimulating its ligase activity [21] (Figure 2A). DDRGK1 and in particular its PCI domain mediates the recruitment of UFM1 cascade components —UFL1, C53/LZAP (CDK5RAP3 also known as CDK5 kinase regulatory subunit-associated protein 3), UFSP2, or ACS1—promoting the formation of enzyme-protein complexes fine tune UFM-1 modification [1][19][21].
Consistent with the theme of bringing together multi-component protein complexes, another player participating in the UFMylation pathway is Cdk5rap3 (Cdk5 regulatory subunit-associated protein 3, C53 or LZAP), a highly conserved protein participating in numerous signaling pathways contributing to tumorigenesis and metastasis which is recruited by DDRGK1 [19][29]. Originally identified in the context of DNA damage and cell cycle control [30][31], CDK5RAP3 has been demonstrated to associate with UFL1 as well as DDRGK1 eliciting its subsequent relocalization to the ER membrane [20][32][33][34]. Moreover, complex formation with UFL1 and DDRGK1 as well as CDK5RAP3, promotes its stability by regulating its degradation [34] as well as affecting overall UFMylation as demonstrated by genetic ablation of CDK5RAP3 in mice [35]. Intriguingly, the presence of CDK5RAP3 seems to be requisite for poly-UFMylation [21][22].
1.4. UFSP1 and UFSP2
UFMylation of substrates is reversed by the action of UFM1-specific cysteine proteases—UFSP1 and UFSP2 [36][37]. Despite having a catalytic triad universally shared by cysteine proteases, UFSP enzymes lack sequence homology to deubiquitinating enzymes (DUBs) or even other proteases, thus representing a novel subfamily of cysteine proteases [37][38]. UFSP1, has a papain-like fold harboring an unusual active site configuration consisting of both a conserved Cys and His box domain as opposed to the classical Cys-His-Asp catalytical triad [37][38] Intriguingly, while murine UFSP1 processes UFM1 effectively, human UFSP1 seems to be catalytically inactive due to the shorter N-terminus and thus lack of the conserved cysteine active site [36][39]. In contrast, crystal structures reveal that murine UFSP2 contains two domains—a C-terminal catalytic domain with a similar architecture as that of murine UFSP1 in addition to a uniquely structured N-terminal domain key to recruitment by DDRGK1 and subsequent relocalization to the ER membrane [17][38]. UFSP2 predominantly resides in the cytoplasm and the nucleus and requires interaction with DDRGK1 for ER membrane recruitment, insinuating that most of its substrates might be in these cellular compartments and ER localization of this protease is necessitated only in specific cellular contexts, i.e., ER stress, UPR etc. [40]. In contrast to the initial observation by Kang et al. [36] that UFSP2 also cleaves the C-terminal amino acids of UFM1, Ishimura et al. demonstrated that knockout of UFSP2 abolished UFM1 deconjugation from its substrates, resulting in the accumulation of UFMylated proteins [11]. Surprisingly, deletion of UFSP2 did not prevent maturation of pro-UFM1, indicating that the cleavage of the C-terminal amino acids serine and cysteine prior to UFM1 activation, might be mediated by currently unidentified UFM1-specific proteases [12][11][21] (Figure 1).
2. Biological Function of UFMylation
Since its discovery more than a decade ago, UFM1 has been linked to diverse cellular processes ranging from hematopoietic cell survival and differentiation during embryogenesis, cell development and tissue homeostasis, transcriptional regulation, mitosis, vesicle trafficking, autophagy, fatty acid metabolism, cellular signaling pathways, and more recently endoplasmic reticulum (ER) homeostasis and DNA damage repair response [1][25][35][41][42][43][44][45]. In contrast to ubiquitination or SUMOylation, UFMylation seems to occur only on several defined substrates [12][20]. Up to date, only a handful of UFM1 substrates including DDRGK1, ASC1, the DNA repair protein MRE11, the ribosomal protein RPL26, ribophorin I (RPN1), and the tumor suppressor protein p53 have been discovered using immunoprecipitation or pulldown approaches (Table 1) [20][21][22][23][25][27][46]. Notwithstanding, the precise molecular mechanisms governing the function of UFM1 on these respective substrates and in the respective cellular pathways are far from being completely understood, as only a handful of UFMylated target proteins have been mechanistically studied up to date (Figure 2).
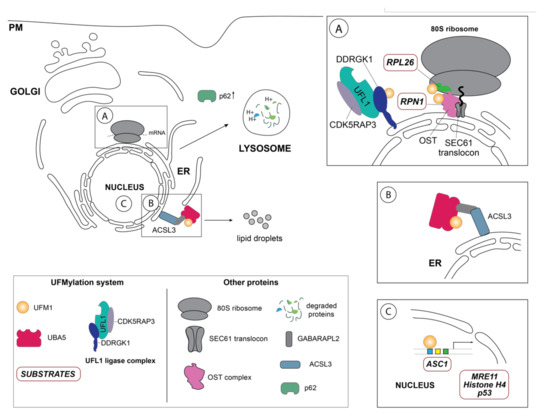
Figure 2. Schematic overview of the so far identified UFM1 substrates and UFM1 enzymes in their respective cellular localization. (A) One of the principal UFM1 substrates—the ribosomal protein RPL26—is UFMylated by the orchestrated action of the UFM1 E3 ligase complex consisting of UFL1, CDK5RAP3 and DDRGK1 and contributes to ERAD as well as an alternative ribosomal quality control mechanism following ribosomal stalling at the SEC61 translocon during co-translational protein targeting and ultimately lysosomal degradation of these aberrant proteins [22][23]. The autophagy cargo adaptor p62 (also sequestosome-1 (SQSTM1) has been found in several studies to indirectly regulate autophagy by UFMylation, although the exact mechanistic details are unclear [47]. Additionally, DDRGK1, an adaptor protein recruiting UFL1 to the ER membrane and modulating its ligase activity, is regulated by UFMylation itself and participates in the degradation of ER sheets [25]. Ribophorin I (RPN1), a protein of the oligosaccharyltransferase (OST) complex, has also been reported to be UFMylated [25]. (B) Inset depicts the GABARAPL2-mediated interaction between the long-chain-fatty-acid CoA ligase 3 (ACSL3) and UBA5, relocalizing it to the ER membrane and stimulating UFMylation [44]. (C) Schematic representation of the nuclear UFMylation substrates, which include the activating signal co-integrator 1 (ASC1), as well as DNA damage repair proteins such as the double strand repair protein MRE11, histone H4, and p53.
2.1. UFM1—A Matter of Survival during Embryonic Development
Genetic deletion of the UFMylation enzymes UBA5, UFL1, as well as the associated adaptor proteins UFBP1 and CDK5RAP3 in both mice and zebrafish revealed the essentiality of UFMylation enzymes for cellular survival and differentiation, in particular that of hematopoietic stem cells [41][48][49]. Moreover, conditional knockout for these genes in mice invoked apoptosis in hematopoietic stem cells (HSCs) through impairment of primitive and definitive erythropoiesis [40][35][49][50], liver cells, and secretory cells in the intestine (Paneth cells) or the pancreas (pancreatic acini), but also concurrently upregulated genes of the UPR or interconnected downstream pathways including inositol-requiring enzyme 1 (IRE1) and X-box binding protein-1 (XBP1), PKR-like ER protein kinase (PERK), and phosphorylated eIF2a [42][51][52]. In particular, germline deletion of UBA5 in mice invoked in severe anemia ultimately leading to embryonic death, while bone marrow of RCAD−/− mice exhibited defective autophagy, as indicated by increased LC3-II and p62 levels [48][50].
Given that CDK5RAP3 functions as an adaptor protein for UFL1 [35], genetic ablation in zebrafish abolished epiboly, which is the first morphogenic change during the gastrulation stage of embryogenesis, most likely through cell-cycle arrest at the G2/M phase of the cell cycle [53][54]. Moreover, analysis of these CDK5RPAP3 deficient zebrafish revealed defective Wnt/β-catenin signaling, which is critical for normal embryonic development. Similarly, while CDK5RAP3 deficient mice were not viable, conditional knockdown of CDK5RAP3 incited severe hypoglycemia and substantially impaired the lipid metabolism ultimately causing lethality [40][35]. Although the CDK5 kinase plays a key role in diverse cellular processes including cell cycle progression, transcriptional regulation, immunity, apoptosis, senescence, cellular differentiation, and DNA damage response [55], it should be noted that the interaction of CDK5RAP3 with UFL1 largely seems to determine these reported phenotypes, rather than just its association with CDK5 kinase [20][34][35][56][57]. However, whether the interaction of CDK5RAP3 with CDK5 kinase modulates UFL1 recruitment and binding remains to be investigated [35][55].
Moreover, in these mouse embryos, CDK5RAP3 deficiency triggered severe anemia as well as major growth retardation in the developing livers, most likely as a consequence of impaired hematopoiesis [35]. In agreement with the observations that genetic ablation of UBA5, the E3-like ligase UFL1 and its adaptor CDK5RAP3 in mice resulted in defective erythropoiesis [35][48][50], germline deletion of DDRGK1, which functions both as a UFL1 adaptor as well as a substrate, evoked aberrant erythroid differentiation [49]. Further study revealed that in particular the primitive and definitive lineages were affected, while loss of UBA5 was lineage specific affected only the erythroid cell lineages [48][50][51]. Unsurprisingly, genetic ablation of UFBP1, similar to that of other components of the UFM1 system, substantially impedes hematopoiesis and ultimately arrests embryogenesis [49]. In line with these observations, conditional knockout of UFBP1 in mice substantially elevated ER stress, repressed transcription of GATA1 and KLF and affected UFMylation of ASC1, which regulates genes modulating erythrocyte development [40][58]. The observation that DDRGK1 and CDK5RAP3 display a differential effect on the erythroid lineage development seems to be reflective of different substrates or a modulated response to UFMylation. Interestingly, UFMylation of the ribosomal protein RPL26 is also differentially influenced by the action of CDK5AP3 and DDRGK1 [22], indicating that these UFL1 adaptors might recruit other proteins that help shape the cellular response to ER stress [22].
2.2. ER Stress: UFM1 to the Rescue
The endoplasmatic reticulum (ER), a dynamic network of membranes is predominantly involved in the assembly and proper folding of proteins in the secretory pathway, which accounts for approximately 30% of the cellular proteome [59][60][61]. To ensure efficiency and fidelity of protein folding, the ER is dynamically regulated by a complex and integrated signal transduction system connecting all components of the secretory pathway. While the ER is constantly adapting to the cellular needs, it becomes essential during ER stress after activation of the unfolded protein response (UPR) to reestablish cellular homeostasis [59][62]. After initiation of the UPR, which utilizes three different signaling pathways (the inositol-requiring transmembrane kinase/endonuclease 1 (IRE1), PKR-like ER protein kinase (PERK), and activating transcription factor 6 (ATF6)), protein translation is halted to allow mRNA translation of UPR proteins, which in turn induce the translation of ER chaperones to upregulate protein folding [59][62]. If this strategy fails to resolve ER stress, the ER network expands and ER-associated degradation (ERAD) of the unfolded or misfolded proteins either by a Ubiquitin-proteasome or by a autophagy-lysosome dependent mechanism as well as upregulating molecular chaperones promoting proper protein folding are initiated [62]. Interestingly, UFM1 was first identified to be transcriptionally upregulated in response to ER stress during the onset and progression of ischemic heart disease in mice [63]. Later, Lemaire et al., observed upregulation of the UFL1 adaptor protein DDRGK1 as well as other components of the UFM1 system upon ER stress induction in the pancreatic acini and the islets of Langerhans [20][64], pointing out the role of UFMylation in secretion [20]. Later, multiple studies also identified the UFL1 adaptor protein DDRGK1 as a UFM1 substrate, requiring UFM1 modification prior to interaction with its cognate ligase [19][20][21][64]. While others have reported UFMylation of DDRGK1 essential to UFL1 ligase function especially during ER stress [21][51], Liang et al., have recently reported that its UFMylation was not required for UFL1 interaction and function during ER-phagy, implying that UFMylation of this adaptor protein might occur only in specific cellular situations and perhaps as a mechanism to regulate its stability or that it might have UFMylation-independent functions [25].
Following the initial observation that the UFM1 ligase UFL1 and its co-factor DDRGK1 modulate ER stress by Lemaire et al., further investigation revealed that transcription of (unfolded protein response) UPR genes such as XBP1 was induced [42] orchestrating the intricate cellular stress response network in a UFM1-dependent manner. Depletion of DDRGK1, which induces moderate ER stress, revealed a regulatory role of the UFM1 system in preserving ER homeostasis through modulation of IREα stability, through its interaction with UFMylated DDRGK1 [51]. Under basal conditions, IREα interacts with BiP (GRP78); however, after UPR activation, it dissociates from BiP, dimerizes and auto-phosphorylates to activate cleavage of the transcription factor XBP-1, which then transcriptionally regulates ER chaperone and ERAD gene expression [65]. Simultaneously, protein expression levels of phosphorylated PERK and BiP (GRP78), key signaling molecules in the UPR-PERK apoptotic pathway, were increased while proteasome inhibition with MG132 prevented Ubiquitin-mediated IREα degradation implying crosstalk with the Ubiquitin Proteasome System [42][51]. Moreover, the intricate interplay between UFMylation and ubiquitination upon disturbance of ER homeostasis becomes evident as genetic disruption of the genes encoding the UFM1 conjugation and deconjugation enzymes, stabilized two soluble luminal ER substrates and integral ER membrane proteins of the HRD1 Ubiquitin ligase [22][66]. In this context, the ubiquitin-dependent degradation of the key UPR signal transducer molecule IREα, which is also a substrate the HRD1 Ubiquitin ligase, is modulated by UFMylation if the interacting protein DDRGK1 [51][65]. Moreover, the action of IREα on the transcription factor XBP-1, introduces an additional regulatory feedback of the UPR response [42][65]. Interestingly, further investigation revealed that impaired UFMylation only had a modest effect on HRD1-dependent ERAD, which may have been causative for the accumulation of misfolded proteins in the ER, as is evidenced by the accumulation of both HRD1 and GRP78 in UFM1 KO cells with ER stress induction [22].
More recently, the repertoire of UFM1 targets has been expanded to ribosomal proteins [22][23][67]. Initially Simsek et al., identified the interaction of UFL1 with the 80S ribosome in a screen for ribosome associating proteins (RAPs) in mouse embryonic fibroblasts as well as the concurrent the UFMylation of three ribosomal subunits—uS3, uS10, uL16, thus implicating a role of UFMylation in embryonic development [67]. Later, Walczak et al., report that RPL26, a ribosomal protein of the large ribosomal 60S subunit located in proximity to the ribosomal tunnel exit is the primary target of UFMylation [22]. Given that UFMylation of RPL26 not only prompts ribosomal ER localization but also appears during translational arrest [23], the authors propose a novel UFMylation-dependent mechanism to resolve translocation-stalled polypeptides and promote the lysosomal clearance of the arrested proteins [23]. In line with this hypothesis, induction of ribosomal stalling by specific inhibitors (i.e., anisomycin) or poly-lysine containing ribosomal stalling constructs (ER_K20), upregulates RPL26 UFMylation thus activating an alternative quality control mechanism distinct from ERAD and cytosolic ribosomal quality control (RQC) to facilitate the lysosomal elimination of these aberrant translation products [68]. Interestingly, the ER stressors, thapsigargin, a sacro/endoplasmatic reticulum Ca2+ ATPase (SERCA) inhibitor, and arsenite induced a weaker UFMylation response than translational inhibitors such as ansiomycin, harringtonine, and cycloheximide, implying that RPL26 UFMylation is prompted by inhibition of ribosomal translation [23].
Later, two independent studies identify the ribosomal protein RPL26 as the primary UFMylation target in mammalian cells upon knockout of the deUFMylase UFSP2 [22][23]. In line with the localization of the UFM1 enzymes UFL1, its adaptor protein DDRGK1 and the deUFMylase UFSP2, UFMylation of RPL26 on its two C-terminal lysine residues (K132 and K134) occurs at the ER membrane compartmentalizing these ribosomes [22][23]. Intriguingly, the UFL1 adaptor protein DDRGK1, which is crucial to promoting the localization of both UFL1 and CDK5RAP3, promotes UFMylation of the lysine 134 of RPL26, while experimental evidence suggests that CDK5RAP3 directs the UFMylation of the second lysine residue [22]. In line with the previous findings, Schuren et al., just recently report that the UFMylation pathway modulates the human cytomegalovirus protein US2-mediated degradation of the HLA class I molecule (HLA-I) by ERAD [69]. Similar to Walczak et al., the authors discovered this interaction through a genome-wide CRISPR screen for US2-mediated HLA-I degradation and later identify UFMylated RPL26 by mass spectrometry in a pulldown from HLA-A2-eGFP expressing cells transduced with STREPII-tagged UFM1 [69]. However, the mechanism of how ribosomal UFMylation affects the dislocation of HLA-I from the ER to the cytosol is lacking and requires further research [69]. While numerous studies have linked UFMylation to ER stress, autophagy, and more recently ER-phagy [20][22][23][25][51], it is intriguing that the UFL1-interacting proteins CDK5RAP3 and DDRGK1 seem to play a major role for mediating these biological processes [22][25][51][56].
2.3. Autophagy—Self Eating for Survival
One of the cellular strategies to alleviate ER stress in addition to UPR is autophagy in which the damaged proteins or organelles are degraded. In particular, macroautophagy of the ER membrane, referred to as ER-phagy, is utilized by the cell to overcome nutrient depletion by the self-eating of the ER membranes [70]. Given the role of UFMylation in maintain ER homeostasis and in participating in the ER stress response, it is unsurprising that UFM1 is involved in ER-phagy as well [25]. These findings consolidate that UFM1 expression levels are upregulated in response to ER stress while perturbation of the UFMylation system induces UPR, thus connecting UFMylation rather unsurprisingly to various diseases including cancer, type 2 diabetes (T2D), cardiovascular diseases, and alcoholic hepatitis [18][20][63][68][71][72][73].
Upon induction of ER stress through various stressors, the UFM1-specific E3 ligase UFL1 mediates UFMylation of its substrate proteins initiating UPR signaling [18]. Failure to resolve ER stress induces autophagy, a highly selective intracellular degradation process, to remove aberrant proteins or damaged organelles, which are recruited by specific receptors to the expanding phagophore [70][74][75]. Upon formation of the double membrane autophagosome and transport to lytic compartments, the contents is then degraded and recycled. Given that the ER participates in the folding and maturation of nearly 30% of the cell’s proteins [60][61], dedicated quality control mechanisms that are interconnected with cellular degradation mechanisms such as autophagy and proteasomal degradation to ensure the fidelity of the process are critical. Thus, to maintain cellular homeostasis through a properly functioning ER, lysosomal degradation of the endoplasmic reticulum—ER-phagy—can be initiated through highly specific cargo receptors [10][76][77]. While it is well established that the Ubiquitin-like modifier LC3 and ATG8 is the crucial to autophagy and ER-phagy, recent reports now indicate that UFM1 is essential to these cellular degradation pathways [56]. Previously, the marked increase of both LC3-II and the cargo receptor p62 in UFL1 depleted murine bone marrow cells attenuating the autophagic flux linked UFMylation to autophagy [50]. However, while the underlying mechanisms of UFM1 participation in autophagy remained unclear, CRISPR screens identified UFMylation not only as a modulator of p62 expression, but also of the autophagy receptors NDP52 and TAXBP1 upon ER stress induction [47]. While these reports describe the effect of UFMylation on the autophagic pathway, they do not provide mechanistic explanation of how the UFM1 system contributes to this process. Discovery of a dual LC3-like and UFM1 interacting (LIR/UFIM) region in the C-terminus of UBA5, which had originally been identified as an GABARAPL2/GATE-16 interactor [12], provides insights into UBA5 recruitment to the ER membrane to activate the UFL1/DDRGK1 E3 complex within in the ER to accomplish substrate UFMylation [4][10]. Recent research demonstrates that the GABARAPL2-mediated association of UBA5 to the ER depended on the lipid droplet biogenesis factor ACSL3 and thereby possibly facilitating UFMylation dynamics [44] (Figure 2B). Interestingly, depletion of ACSL3 downregulated UBA5, DDRGK1 and UFL1 transcription, suggesting that perhaps transcriptional regulation of UFMylation occurs during lipid droplet biogenesis, which is induced during ER stress and is essential for ER-phagy [25][78]. Just recently, C53 (CDK5RAP3), was identified in an immunoprecipitation-mass spec (IP-MS) screen as a receptor for proteins destined for autophagic degradation [56]. Further mechanistic dissection revealed that C53 interacted upon stalled proteins upon ribosomal stalling during co-translational protein translocation and its depletion diminished autophagic degradation of these aberrant proteins [56].
Corroborating these observations, Jiang et al., report that DDRGK1-mediated UFMylation of RPL26, which mediates the degradation of ER sheets, as well as of the quality-control factor ribophorin 1 (RPN1) glycosylating faulty ER proteins during co-translational translocation [25][77]. Perhaps unsurprisingly in the context of ER-phagy, numerous proteins involved in endocytic pathway the RAB-GTPases RAB1A/RAB5C, Clathrin, and the ADP-ribosylation factor ARF4 were identified to be UFMylated in a DDRGK1-dependent manner [25]. Earlier, Zhang et al., found that perturbation of ER homeostasis but also inhibition of vesicle trafficking with Brefeldin A upregulated the expression of UFM1, UBA5, UFL1 and C53, which could be reversed by depletion of XBP-1 [42]. In line with these observations, UFC1 and UFM1, were identified to interact with the neural adhesion molecule NCAM 140 (also known as NCAM1, isoform 2 or CD56) promoting its endocytosis, implicating UFMylation in the endocytic pathway [79]. In addition to regulating ER homeostasis primarily through post-translational modification as well as through protein interaction with DDRGK1, UFM1 has been implicated in transcriptional control of several genes involved in the cellular UPR. Most notably, p62 (SQSTM) and XBP1 have been demonstrated to be transcriptionally modulated by UFMylation introducing an additional layer of ER homeostasis maintenance [42][47]. Genetic depletion of UFM1 induced CHOP and SQSTM expression, while Zhang et al. report that UFM1 binds to XBP-1, a transcription factor crucial to initiating the UPR response as well as a plethora of dedicated transcriptional networks regulating cell-type and condition-specific responses, in ChIP and luciferase assays [42].
2.4. Transcriptional Regulation
While UFM1 has been shown to transcriptionally regulate UPR to restore cellular homeostasis [42][51], it has also been linked to transactivation of estrogen receptor α (ERα) as well as in modulating expression levels of genes critical for erythropoiesis [41][49]. Moreover, the transcriptional activation of activating signal co-integrator 1 (ASC1) is induced by UFMylation to promote estrogen receptor α (ERα) transactivation [21]. One of the few identified targets is the activating signal co-integrator (ASC1), a known co-activator of the estrogen receptor ERα, was discovered to be UFMylated in a DDRGK1 dependent manner at four different lysine acceptor sites (K234, K325, K334 and K367) [21]. Interestingly, UFMylation of ASC1 occurred only upon stimulation with estrogen (E2), which displaced UFSP2 bound to ASC1, enlisting UFL1 and DDRGK1 to promote poly-UFM1 chain formation by utilizing the internal lysine (K69) of UFM1 [21]. UFMylation of ASC1 is key to promoting ERα transactivation as well as transcriptional activation of ERα target genes such as pS2, cyclin D1, and c-Myc [21]. These findings indicate that UFMylation of ASC1 might mediate tumor formation and growth in ERα-dependent breast cancer [21]. Correspondently, Zhang et al., observed that the expression levels of genes regulating erythroid lineage (ε-globin, βH1-globin) and the cognate transcription factors (GATA-1/FOG-1 and KLF) were markedly reduced in UFL1 deficient fetal livers, thus assigning UFM1 a role in transcription [50]. In addition to partaking in transcriptional regulation of a variety of genes [80], the long non-coding RNAs (lncRNA) of UFC1, which are large RNA transcripts that do not encode proteins but have regulatory roles [80][81], have been found to critically modulate oncogene expression thus contributing to tumorigenesis, progression and metastasis, in numerous cancers [82][83].
2.5. Signaling Pathways
Corroborating its role in modulating transcription, UFM1 has been implicated in orchestrating cellular signal transduction pathways such as the NF-kB and JNK signaling cascades [71][84][85]. Initial studies described that both UFL1 and the adaptor protein C53 (CDK5RAP3), reported to function as a tumor suppressor [30][86][87][88], regulates NF-kB signaling by associating directly with RELA (p65), abolishing its phosphorylation to enhance interaction with HDAC thereby stimulating NF-κb activity [34]. Later, repression of liposaccharide (LPS) induced nuclear translocation of NF-κb, which initiates its transcriptional activation as well as transcription of inflammatory genes such as TNF-α, IL-6, IL-1β, and IL-12, was found to be modulated by UFM1 system [72][84][89]. Another example of the role of UFM1 in coordinating cellular signal transduction is the UFMylation of ASC1 upon ERα binding promoting the ERα transactivation to modulate expression of ERα target genes including Cyclin D1 and c-Myc [21]. In the light of these findings, it becomes clear that UFMylation is an important player in signal transduction, however many details and the extent of its participation remain to be understood.
2.6. UFMylation and the DNA Damage Response
Upon insult to the genome leading to DNA double strand breaks (DSBs), expeditious DNA damage response (DDR) is required to maintain genomic integrity and is tightly regulated by a host of post-translational modifications including phosphorylation, methylation, acetylation, ubiquitination, and more recently UFMylation [90]. Initiation of DDR occurs through activation of the MRE11-RAD50-NBS1 (MRN) complex subsequent to histone H2AX phosphorylation at the DNA damage sites mediated by ataxia-telangiectasia mutated (ATM) kinase leading to DNA repair protein recruitment by the MRN complex [91]. Moreover, further regulation of ATM is introduced by the interaction of the acetyltransferase Tip60 with tri-methylated histone H3 (H3K9me3) which then induces ATM acteylation thus amplifying its activation through its autophosphorylation [91]. However, UFL1-mediated UFMylation of MRE11 on lysine 282 was found to be critical for MRN complex formation, immediate recruitment to DNA damage sites, as well as for proper ATM activation to initiate DNA repair [27]. Prior to activation and recruitment of the MRN complex to the DNA double strand breaks (DSBs), ATM kinase phosphorylates UFL1, an interactor of the MRN complex, enhancing its ligase activity and inciting UFMylation of histone H4 in a Tip60-dependent manner, providing a positive feedback loop for ATM activation [26]. Furthermore, Qin et al., found that UFL1-mediated histone H4 mono-UFMylation, coordinates Tip60 and Suv39h1 recruitment in a serine/threonine kinase 38 (STK38) dependent fashion and mediates subsequent histone H3 trimethylation (H3K9me3) to synchronize the DDR [26][92]. Just recently, the tumor suppressor p53, which is post-translationally modified by phosphorylation, acetylation, SUMOylation, NEDDylation, and ubiquitination orchestrating a differential cellular response to a plethora of cellular stimuli [93], was reported to be UFMylated at four lysine residues in C-terminal domain intrinsically suppressing tumorigenesis [46]. UFMylation at this PTM hotspot, possibly prevents MDM2 ligase recruitment and subsequent ubiquitination thereby antagonizing p53 degradation. Despite this finding, further research needs to be undertaken to investigate the crosstalk between UFMylation and other PTMs on p53 in response to cellular stressors or DNA damage.
In the light of earlier findings that UFM1 modification is necessitated for timely DNA damage response upon double strand breaks [94][26][27][92], UFMylation of p53 could potentially represent a signalling pathway leading to the rapid MRN complex recruitment and concomitant activation of ATM kinase [26][27][46]. The participation of UFM1 in the early events of the DNA damage response pathway as well as its role in modulating p53 function, links the Ubiquitin-like modifier with disease onset, in particular with tumorigenesis and cancer progression.
This entry is adapted from the peer-reviewed paper 10.3390/biom11020255
References
- Wei, Y.; Xu, X. UFMylation: A Unique & Fashionable Modification for Life. Genom. Proteom. Bioinform. 2016, 14, 140–146.
- Schulman, B.A.; Harper, J.W. Ubiquitin-like protein activation by E1 enzymes: The apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 2009, 10, 319–331.
- Padala, P.; Oweis, W.; Mashahreh, B.; Soudah, N.; Cohen-Kfir, E.; Todd, E.A.; Berndsen, C.E.; Wiener, R. Novel insights into the interaction of UBA5 with UFM1 via a UFM1-interacting sequence. Sci. Rep. 2017, 7, 508.
- Habisov, S.; Huber, J.; Ichimura, Y.; Akutsu, M.; Rogova, N.; Loehr, F.; McEwan, D.G.; Johansen, T.; Dikic, I.; Doetsch, V.; et al. Structural and Functional Analysis of a Novel Interaction Motif within UFM1-activating Enzyme 5 (UBA5) Required for Binding to Ubiquitin-like Proteins and Ufmylation. J. Biol. Chem. 2016, 291, 9025–9041.
- Soudah, N.; Padala, P.; Hassouna, F.; Kumar, M.; Mashahreh, B.; Lebedev, A.A.; Isupov, M.N.; Cohen-Kfir, E.; Wiener, R. An N-Terminal Extension to UBA5 Adenylation Domain Boosts UFM1 Activation: Isoform-Specific Differences in Ubiquitin-like Protein Activation. J. Mol. Biol. 2019, 431, 463–478.
- Lv, Z.; Olsen, S.K. UFM1-Activating Enzyme 5 (Uba5) Requires an Extension to Get the Job Done Right. J. Mol. Biol. 2019, 431, 479–482.
- Bacik, J.P.; Walker, J.R.; Ali, M.; Schimmer, A.D.; Dhe-Paganon, S. Crystal structure of the human ubiquitin-activating enzyme 5 (UBA5) bound to ATP: Mechanistic insights into a minimalistic E1 enzyme. J. Biol. Chem. 2010, 285, 20273–20280.
- Gavin, J.M.; Hoar, K.; Xu, Q.; Ma, J.; Lin, Y.; Chen, J.; Chen, W.; Bruzzese, F.J.; Harrison, S.; Mallender, W.D.; et al. Mechanistic study of Uba5 enzyme and the Ufm1 conjugation pathway. J. Biol. Chem. 2014, 289, 22648–22658.
- Mashahreh, B.; Hassouna, F.; Soudah, N.; Cohen-Kfir, E.; Strulovich, R.; Haitin, Y.; Wiener, R. Trans-binding of UFM1 to UBA5 stimulates UBA5 homodimerization and ATP binding. FASEB J. 2018, 32, 2794–2802.
- Huber, J.; Obata, M.; Gruber, J.; Akutsu, M.; Lohr, F.; Rogova, N.; Guntert, P.; Dikic, I.; Kirkin, V.; Komatsu, M.; et al. An atypical LIR motif within UBA5 (ubiquitin like modifier activating enzyme 5) interacts with GABARAP proteins and mediates membrane localization of UBA5. Autophagy 2020, 16, 256–270.
- Ishimura, R.; Obata, M.; Kageyama, S.; Daniel, J.; Tanaka, K.; Komatsu, M. A novel approach to assess the ubiquitin-fold modifier 1-system in cells. FEBS Lett. 2017, 591, 196–204.
- Komatsu, M.; Chiba, T.; Tatsumi, K.; Iemura, S.; Tanida, I.; Okazaki, N.; Ueno, T.; Kominami, E.; Natsume, T.; Tanaka, K. A novel protein-conjugating system for Ufm1, a ubiquitin-fold modifier. EMBO J. 2004, 23, 1977–1986.
- Mizushima, T.; Tatsumi, K.; Ozaki, Y.; Kawakami, T.; Suzuki, A.; Ogasahara, K.; Komatsu, M.; Kominami, E.; Tanaka, K.; Yamane, T. Crystal structure of Ufc1, the Ufm1-conjugating enzyme. Biochem. Biophys. Res. Commun. 2007, 362, 1079–1084.
- Liu, G.; Aramini, J.; Atreya, H.S.; Eletsky, A.; Xiao, R.; Acton, T.; Ma, L.; Montelione, G.T.; Szyperski, T. GFT NMR based resonance assignment for the 21 kDa human protein UFC1. J. Biomol. NMR 2005, 32, 261.
- Liu, G.; Forouhar, F.; Eletsky, A.; Atreya, H.S.; Aramini, J.M.; Xiao, R.; Huang, Y.J.; Abashidze, M.; Seetharaman, J.; Liu, J.; et al. NMR and X-RAY structures of human E2-like ubiquitin-fold modifier conjugating enzyme 1 (UFC1) reveal structural and functional conservation in the metazoan UFM1-UBA5-UFC1 ubiquination pathway. J. Struct. Funct. Genom. 2009, 10, 127–136.
- Xie, S. Characterization, crystallization and preliminary X-ray crystallographic analysis of the human Uba5 C-terminus-Ufc1 complex. Acta Crystallogr. F Struct. Biol. Commun. 2014, 70, 1093–1097.
- Daniel, J.; Liebau, E. The ufm1 cascade. Cells 2014, 3, 627–638.
- Xie, Z.; Fang, Z.; Pan, Z. Ufl1/RCAD, a Ufm1 E3 ligase, has an intricate connection with ER stress. Int. J. Biol. Macromol. 2019, 135, 760–767.
- Tatsumi, K.; Sou, Y.S.; Tada, N.; Nakamura, E.; Iemura, S.; Natsume, T.; Kang, S.H.; Chung, C.H.; Kasahara, M.; Kominami, E.; et al. A novel type of E3 ligase for the Ufm1 conjugation system. J. Biol. Chem. 2010, 285, 5417–5427.
- Lemaire, K.; Moura, R.F.; Granvik, M.; Igoillo-Esteve, M.; Hohmeier, H.E.; Hendrickx, N.; Newgard, C.B.; Waelkens, E.; Cnop, M.; Schuit, F. Ubiquitin fold modifier 1 (UFM1) and its target UFBP1 protect pancreatic beta cells from ER stress-induced apoptosis. PLoS ONE 2011, 6, e18517.
- Yoo, H.M.; Kang, S.H.; Kim, J.Y.; Lee, J.E.; Seong, M.W.; Lee, S.W.; Ka, S.H.; Sou, Y.S.; Komatsu, M.; Tanaka, K.; et al. Modification of ASC1 by UFM1 is crucial for ERalpha transactivation and breast cancer development. Mol. Cell 2014, 56, 261–274.
- Walczak, C.P.; Leto, D.E.; Zhang, L.; Riepe, C.; Muller, R.Y.; DaRosa, P.A.; Ingolia, N.T.; Elias, J.E.; Kopito, R.R. Ribosomal protein RPL26 is the principal target of UFMylation. Proc. Natl. Acad. Sci. USA 2019, 116, 1299–1308.
- Wang, L.; Xu, Y.; Rogers, H.; Saidi, L.; Noguchi, C.T.; Li, H.; Yewdell, J.W.; Guydosh, N.R.; Ye, Y. UFMylation of RPL26 links translocation-associated quality control to endoplasmic reticulum protein homeostasis. Cell Res. 2020, 30, 5–20.
- Scheel, H.; Hofmann, K. Prediction of a common structural scaffold for proteasome lid, COP9-signalosome and eIF3 complexes. BMC Bioinform. 2005, 6, 71.
- Liang, J.R.; Lingeman, E.; Luong, T.; Ahmed, S.; Muhar, M.; Nguyen, T.; Olzmann, J.A.; Corn, J.E. A Genome-wide ER-phagy Screen Highlights Key Roles of Mitochondrial Metabolism and ER-Resident UFMylation. Cell 2020, 180, 1160–1177.
- Qin, B.; Yu, J.; Nowsheen, S.; Wang, M.; Tu, X.; Liu, T.; Li, H.; Wang, L.; Lou, Z. UFL1 promotes histone H4 ufmylation and ATM activation. Nat. Commun. 2019, 10, 1242.
- Wang, Z.; Gong, Y.; Peng, B.; Shi, R.; Fan, D.; Zhao, H.; Zhu, M.; Zhang, H.; Lou, Z.; Zhou, J.; et al. MRE11 UFMylation promotes ATM activation. Nucleic Acids Res. 2019, 47, 4124–4135.
- Duda, D.M.; Borg, L.A.; Scott, D.C.; Hunt, H.W.; Hammel, M.; Schulman, B.A. Structural insights into NEDD8 activation of cullin-RING ligases: Conformational control of conjugation. Cell 2008, 134, 995–1006.
- Mak, G.W.; Lai, W.L.; Zhou, Y.; Li, M.; Ng, I.O.; Ching, Y.P. CDK5RAP3 is a novel repressor of p14ARF in hepatocellular carcinoma cells. PLoS ONE 2012, 7, e42210.
- Jiang, H.; Wu, J.; He, C.; Yang, W.; Li, H. Tumor suppressor protein C53 antagonizes checkpoint kinases to promote cyclin-dependent kinase 1 activation. Cell Res. 2009, 19, 458–468.
- Jiang, H.; Luo, S.; Li, H. Cdk5 activator-binding protein C53 regulates apoptosis induced by genotoxic stress via modulating the G2/M DNA damage checkpoint. J. Biol. Chem. 2005, 280, 20651–20659.
- Kwon, J.; Cho, H.J.; Han, S.H.; No, J.G.; Kwon, J.Y.; Kim, H. A novel LZAP-binding protein, NLBP, inhibits cell invasion. J. Biol. Chem. 2010, 285, 12232–12240.
- Shiwaku, H.; Yoshimura, N.; Tamura, T.; Sone, M.; Ogishima, S.; Watase, K.; Tagawa, K.; Okazawa, H. Suppression of the novel ER protein Maxer by mutant ataxin-1 in Bergman glia contributes to non-cell-autonomous toxicity. EMBO J. 2010, 29, 2446–2460.
- Wu, J.; Lei, G.; Mei, M.; Tang, Y.; Li, H. A novel C53/LZAP-interacting protein regulates stability of C53/LZAP and DDRGK domain-containing Protein 1 (DDRGK1) and modulates NF-kappaB signaling. J. Biol. Chem. 2010, 285, 15126–15136.
- Yang, R.; Wang, H.; Kang, B.; Chen, B.; Shi, Y.; Yang, S.; Sun, L.; Liu, Y.; Xiao, W.; Zhang, T.; et al. CDK5RAP3, a UFL1 substrate adaptor, is crucial for liver development. Development 2019, 146, dev169235.
- Kang, S.H.; Kim, G.R.; Seong, M.; Baek, S.H.; Seol, J.H.; Bang, O.S.; Ovaa, H.; Tatsumi, K.; Komatsu, M.; Tanaka, K.; et al. Two novel ubiquitin-fold modifier 1 (Ufm1)-specific proteases, UfSP1 and UfSP2. J. Biol. Chem. 2007, 282, 5256–5262.
- Ha, B.H.; Ahn, H.C.; Kang, S.H.; Tanaka, K.; Chung, C.H.; Kim, E.E. Structural basis for Ufm1 processing by UfSP1. J. Biol. Chem. 2008, 283, 14893–14900.
- Ha, B.H.; Jeon, Y.J.; Shin, S.C.; Tatsumi, K.; Komatsu, M.; Tanaka, K.; Watson, C.M.; Wallis, G.; Chung, C.H.; Kim, E.E. Structure of ubiquitin-fold modifier 1-specific protease UfSP2. J. Biol. Chem. 2011, 286, 10248–10257.
- Witting, K.F.; van Noort, G.J.v.d.H.; Kofoed, C.; Ormeno, C.T.; Atmioui, D.E.; Mulder, M.P.C.; Ovaa, H. Generation of the UFM1 Toolkit for Profiling UFM1-Specific Proteases and Ligases. Angew. Chem. Int. Ed. Engl. 2018, 57, 14164–14168.
- Gerakis, Y.; Quintero, M.; Li, H.; Hetz, C. The UFMylation System in Proteostasis and Beyond. Trends Cell Biol. 2019, 29, 974–986.
- Cai, Y.; Singh, N.; Li, H. Essential role of Ufm1 conjugation in the hematopoietic system. Exp. Hematol. 2016, 44, 442–446.
- Zhang, Y.; Zhang, M.; Wu, J.; Lei, G.; Li, H. Transcriptional regulation of the Ufm1 conjugation system in response to disturbance of the endoplasmic reticulum homeostasis and inhibition of vesicle trafficking. PLoS ONE 2012, 7, e48587.
- Merbl, Y.; Refour, P.; Patel, H.; Springer, M.; Kirschner, M.W. Profiling of ubiquitin-like modifications reveals features of mitotic control. Cell 2013, 152, 1160–1172.
- Eck, F.; Phuyal, S.; Smith, M.D.; Kaulich, M.; Wilkinson, S.; Farhan, H.; Behrends, C. ACSL3 is a novel GABARAPL2 interactor that links ufmylation and lipid droplet biogenesis. J. Cell Sci. 2020, 133, jcs243477.
- Lin, J.X.; Xie, X.S.; Weng, X.F.; Qiu, S.L.; Yoon, C.; Lian, N.Z.; Xie, J.W.; Wang, J.B.; Lu, J.; Chen, Q.Y.; et al. UFM1 suppresses invasive activities of gastric cancer cells by attenuating the expres7sion of PDK1 through PI3K/AKT signaling. J. Exp. Clin. Cancer Res. 2019, 38, 410.
- Liu, J.; Guan, D.; Dong, M.; Yang, J.; Wei, H.; Liang, Q.; Song, L.; Xu, L.; Bai, J.; Liu, C.; et al. UFMylation maintains tumour suppressor p53 stability by antagonizing its ubiquitination. Nat. Cell Biol. 2020, 22, 1056–1063.
- De Jesus, R.; Moretti, F.; McAllister, G.; Wang, Z.; Bergman, P.; Liu, S.; Frias, E.; Alford, J.; Reece-Hoyes, J.S.; Lindeman, A.; et al. Functional CRISPR screening identifies the ufmylation pathway as a regulator of SQSTM1/p62. Elife 2016, 5, e17290.
- Tatsumi, K.; Yamamoto-Mukai, H.; Shimizu, R.; Waguri, S.; Sou, Y.S.; Sakamoto, A.; Taya, C.; Shitara, H.; Hara, T.; Chung, C.H.; et al. The Ufm1-activating enzyme Uba5 is indispensable for erythroid differentiation in mice. Nat. Commun. 2011, 2, 181.
- Cai, Y.; Pi, W.; Sivaprakasam, S.; Zhu, X.; Zhang, M.; Chen, J.; Makala, L.; Lu, C.; Wu, J.; Teng, Y.; et al. UFBP1, a Key Component of the Ufm1 Conjugation System, Is Essential for Ufmylation-Mediated Regulation of Erythroid Development. PLoS Genet. 2015, 11, e1005643.
- Zhang, M.; Zhu, X.; Zhang, Y.; Cai, Y.; Chen, J.; Sivaprakasam, S.; Gurav, A.; Pi, W.; Makala, L.; Wu, J.; et al. RCAD/Ufl1, a Ufm1 E3 ligase, is essential for hematopoietic stem cell function and murine hematopoiesis. Cell Death Differ. 2015, 22, 1922–1934.
- Liu, J.; Wang, Y.; Song, L.; Zeng, L.; Yi, W.; Liu, T.; Chen, H.; Wang, M.; Ju, Z.; Cong, Y.S. A critical role of DDRGK1 in endoplasmic reticulum homoeostasis via regulation of IRE1alpha stability. Nat. Commun. 2017, 8, 14186.
- Cai, Y.; Zhu, G.; Liu, S.; Pan, Z.; Quintero, M.; Poole, C.J.; Lu, C.; Zhu, H.; Islam, B.; Riggelen, J.V.; et al. Indispensable role of the Ubiquitin-fold modifier 1-specific E3 ligase in maintaining intestinal homeostasis and controlling gut inflammation. Cell Discov. 2019, 5, 7.
- Lin, K.Y.; Kao, S.H.; Lai, C.M.; Chen, C.T.; Wu, C.Y.; Hsu, H.J.; Wang, W.D. Tumor Suppressor Lzap Suppresses Wnt/beta-Catenin Signaling to Promote Zebrafish Embryonic Ventral Cell Fates via the Suppression of Inhibitory Phosphorylation of Glycogen Synthase Kinase 3. J. Biol. Chem. 2015, 290, 29808–29819.
- Bruce, A.E.E.; Heisenberg, C.P. Mechanisms of zebrafish epiboly: A current view. Curr. Top. Dev. Biol. 2020, 136, 319–341.
- Sharma, S.; Sicinski, P. A kinase of many talents: Non-neuronal functions of CDK5 in development and disease. Open Biol. 2020, 10, 190287.
- Stephani, M.; Picchianti, L.; Dagdas, Y. C53 is a cross-kingdom conserved reticulophagy receptor that bridges the gap betweenselective autophagy and ribosome stalling at the endoplasmic reticulum. Autophagy 2020, 1–2.
- Ching, Y.P.; Qi, Z.; Wang, J.H. Cloning of three novel neuronal Cdk5 activator binding proteins. Gene 2000, 242, 285–294.
- Neziri, D.; Ilhan, A.; Maj, M.; Majdic, O.; Baumgartner-Parzer, S.; Cohen, G.; Base, W.; Wagner, L. Cloning and molecular characterization of Dashurin encoded by C20orf116, a PCI-domain containing protein. Biochim. Biophys. Acta 2010, 1800, 430–438.
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102.
- Chen, X.; Karnovsky, A.; Sans, M.D.; Andrews, P.C.; Williams, J.A. Molecular characterization of the endoplasmic reticulum: Insights from proteomic studies. Proteomics 2010, 10, 4040–4052.
- Chen, Y.; Zhang, Y.; Yin, Y.; Gao, G.; Li, S.; Jiang, Y.; Gu, X.; Luo, J. SPD—A web-based secreted protein database. Nucleic Acids Res. 2005, 33, D169–D173.
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977.
- Azfer, A.; Niu, J.; Rogers, L.M.; Adamski, F.M.; Kolattukudy, P.E. Activation of endoplasmic reticulum stress response during the development of ischemic heart disease. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1411–H1420.
- Zhu, Y.; Lei, Q.; Li, D.; Zhang, Y.; Jiang, X.; Hu, Z.; Xu, G. Proteomic and Biochemical Analyses Reveal a Novel Mechanism for Promoting Protein Ubiquitination and Degradation by UFBP1, a Key Component of Ufmylation. J. Proteome Res. 2018, 17, 1509–1520.
- Sun, S.; Shi, G.; Sha, H.; Ji, Y.; Han, X.; Shu, X.; Ma, H.; Inoue, T.; Gao, B.; Kim, H.; et al. IRE1alpha is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat. Cell Biol. 2015, 17, 1546–1555.
- Bagola, K.; Mehnert, M.; Jarosch, E.; Sommer, T. Protein dislocation from the ER. Biochim. Biophys. Acta 2011, 1808, 925–936.
- Simsek, D.; Tiu, G.C.; Flynn, R.A.; Byeon, G.W.; Leppek, K.; Xu, A.F.; Chang, H.Y.; Barna, M. The Mammalian Ribo-interactome Reveals Ribosome Functional Diversity and Heterogeneity. Cell 2017, 169, 1051–1065.
- Wang, S.; Jia, M.; Su, M.; Hu, X.; Li, J.; Xu, Y.; Qiu, W. Ufmylation Is Activated in Renal Cancer and Is Not Associated with von Hippel-Lindau Mutation. DNA Cell Biol. 2020, 39, 654–660.
- Schuren, A.B.C.; Boer, I.G.J.; Bouma, E.M.; Van de Weijer, M.L.; Costa, A.I.; Hubel, P.; Pichlmair, A.; Lebbink, R.J.; Wiertz, E. The UFM1 Pathway Impacts HCMV US2-Mediated Degradation of HLA Class I. Molecules 2021, 26, 287.
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364.
- Hu, X.; Zhang, H.; Song, Y.; Zhuang, L.; Yang, Q.; Pan, M.; Chen, F. Ubiquitin fold modifier 1 activates NF-kappaB pathway by down-regulating LZAP expression in the macrophage of diabetic mouse model. Biosci. Rep. 2020, 40, BSR20191672.
- Li, J.; Yue, G.; Ma, W.; Zhang, A.; Zou, J.; Cai, Y.; Tang, X.; Wang, J.; Liu, J.; Li, H.; et al. Ufm1-Specific Ligase Ufl1 Regulates Endoplasmic Reticulum Homeostasis and Protects Against Heart Failure. Circ. Heart Fail. 2018, 11, e004917.
- Miller, C.; Cai, Y.; Patton, T.; Graves, S.H.; Li, H.; Sabbatini, M.E. RCAD/BiP pathway is necessary for the proper synthesis of digestive enzymes and secretory function of the exocrine pancreas. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G314–G326.
- Johansen, T.; Lamark, T. Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. J. Mol. Biol. 2020, 432, 80–103.
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501.
- Song, S.; Tan, J.; Miao, Y.; Zhang, Q. Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement of UPR and the core autophagy machinery. J. Cell Physiol. 2018, 233, 3867–3874.
- Strzyz, P. Foundations of ER-phagy regulation. Nat. Rev. Mol. Cell Biol. 2020, 21, 251.
- Jarc, E.; Petan, T. Lipid Droplets and the Management of Cellular Stress. Yale J. Biol. Med. 2019, 92, 435–452.
- Homrich, M.; Wobst, H.; Laurini, C.; Sabrowski, J.; Schmitz, B.; Diestel, S. Cytoplasmic domain of NCAM140 interacts with ubiquitin-fold modifier-conjugating enzyme-1 (Ufc1). Exp. Cell Res. 2014, 324, 192–199.
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2020, 22, 96–118.
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208.
- Yu, T.; Shan, T.D.; Li, J.Y.; Huang, C.Z.; Wang, S.Y.; Ouyang, H.; Lu, X.J.; Xu, J.H.; Zhong, W.; Chen, Q.K. Knockdown of linc-UFC1 suppresses proliferation and induces apoptosis of colorectal cancer. Cell Death Dis. 2016, 7, e2228.
- Beckedorff, F.C.; Amaral, M.S.; Deocesano-Pereira, C.; Verjovski-Almeida, S. Long non-coding RNAs and their implications in cancer epigenetics. Biosci. Rep. 2013, 33, 54.
- Li, Y.Y.; Zhang, G.Y.; He, J.P.; Zhang, D.D.; Kong, X.X.; Yuan, H.M.; Chen, F.L. Ufm1 inhibits LPS-induced endothelial cell inflammatory responses through the NF-kappaB signaling pathway. Int. J. Mol. Med. 2017, 39, 1119–1126.
- Kuang, M.; Yang, M.; Li, L.; Li, C.; Wang, G. UFM1-Specific Ligase 1 Ligating Enzyme 1 Mediates Milk Protein and Fat Synthesis-Related Gene Expression via the JNK Signaling Pathway in Mouse Mammary Epithelial Cells. Oxid. Med. Cell Longev. 2020, 2020, 4045674.
- Lin, J.X.; Yoon, C.; Li, P.; Ryeom, S.W.; Cho, S.J.; Zheng, C.H.; Xie, J.W.; Wang, J.B.; Lu, J.; Chen, Q.Y.; et al. CDK5RAP3 as tumour suppressor negatively regulates self-renewal and invasion and is regulated by ERK1/2 signalling in human gastric cancer. Br. J. Cancer 2020, 123, 1131–1144.
- Chen, Q.Y.; Liu, L.C.; Wang, J.B.; Xie, J.W.; Lin, J.X.; Lu, J.; Cao, L.L.; Lin, M.; Tu, R.H.; Huang, C.M.; et al. CDK5RAP3 Inhibits the Translocation of MCM6 to Influence the Prognosis in Gastric Cancer. J. Cancer 2019, 10, 4488–4498.
- Lin, J.X.; Xie, X.S.; Weng, X.F.; Zheng, C.H.; Xie, J.W.; Wang, J.B.; Lu, J.; Chen, Q.Y.; Cao, L.L.; Lin, M.; et al. Low expression of CDK5RAP3 and DDRGK1 indicates a poor prognosis in patients with gastric cancer. World J. Gastroenterol. 2018, 24, 3898–3907.
- Yang, G.; Wang, Y.; Chen, Y.; Huang, R. UFL1 attenuates IL-1beta-induced inflammatory response in human osteoarthritis chondrocytes. Int. Immunopharmacol. 2020, 81, 106278.
- Harper, J.W.; Elledge, S.J. The DNA damage response: Ten years after. Mol. Cell 2007, 28, 739–745.
- Lamarche, B.J.; Orazio, N.I.; Weitzman, M.D. The MRN complex in double-strand break repair and telomere maintenance. FEBS Lett. 2010, 584, 3682–3695.
- Qin, B.; Yu, J.; Nowsheen, S.; Zhao, F.; Wang, L.; Lou, Z. STK38 promotes ATM activation by acting as a reader of histone H4 ufmylation. Sci. Adv. 2020, 6, eaax8214.
- Dai, C.; Gu, W. p53 post-translational modification: Deregulated in tumorigenesis. Trends Mol. Med. 2010, 16, 528–536.
- Fang, Z.; Pan, Z. Essential Role of Ubiquitin-Fold Modifier 1 Conjugation in DNA Damage Response. DNA Cell Biol. 2019, 38, 1030–1039.