Tomato (Solanum lycopersicum L.) is a model system for studying the molecular basis of resistance in plants. The investigation of evolutionary dynamics of tomato resistance (R)-loci provides unique opportunities for identifying factors that promote or constrain genome evolution. Nucleotide-binding domain and leucine-rich repeat (NB-LRR) receptors belong to one of the most plastic and diversified families. The vast amount of genomic data available for Solanaceae and wild tomato relatives provides unprecedented insights into the patterns and mechanisms of evolution of NB-LRR genes. Comparative analysis remarked a reshuffling of R-islands on chromosomes and a high degree of adaptive diversification in key R-loci induced by species-specific pathogen pressure.
- R-genes
- gene clusters
- NLR genes
- evolutionary dynamics
- genomic-driven breeding
1. Introduction
Tomato (Solanum lycopersicum L.) belongs to the large and diverse Solanaceae family, also referred to as nightshade. Species in the Solanum section Lycopersicon originated in the region that extends from Andean Highlands to the coast of Galapagos islands and includes the domesticated tomato and its 12 closest wild relatives (S. arcanum, S. cheesmaniae, S. chilense, S. chmielewskii, S. corneliomulleri, S. galapagense, S. habrochaites, S. huaylasense, S. neorickii, S. pennellii, S. peruvianum and S. pimpinellifolium) [1].
The economical and nutritional importance places tomato among the most widely studied crops, thus making it a model to understand the molecular processes related to plant–pathogen interaction [2]. A wide range of biotic stresses impairs tomato yield, most of which have been largely investigated to better understand which molecular mechanisms are activated during diseases [3].
The tomato genome sequence has been released over eight years ago [4], totally revolutionizing the pace of breeding activities. Scientists and breeders around the world actively use the genome to investigate the tomato “sequence space” with different goals. The first exhaustive annotation of tomato R-genes was released six years ago [5], and a tomato R-gene functional map was published immediately after [6].
Wild tomato species are characterized by a wide genetic variability as they occupy different habitats along a diversified climatic gradient [7]. By contrast, cultivated tomato has faced several bottlenecks during its domestication history; this led to a drastic reduction of its genetic diversity [8,9]. Therefore, it is necessary to recover the untapped variability of wild tomato relatives, as they represent the primary source of resistance for the cultivated tomato, being rich in genes conferring resistance to a large panel of pathogens [10]. The identification of suitable sources of new resistance is one of the most straightforward strategies for obtaining pathogen-resistant tomato varieties. Therefore, the identification and characterization of resistance (R)-genes in tomato wild relatives is definitely useful for both classical and innovative breeding strategies.
As the amount of sequenced genomes increases, comparative genomics is becoming an even more powerful method for identifying functionally important loci [11]. Comparative approaches across Solanaceae species have proved particularly useful for gaining new insights into the evolution and functional diversification of R-genes. Investigations on R-gene families in other Solanaceae species (potato, pepper, and eggplant) have helped to consolidate our knowledge on the processes that mediate disease resistance/tolerance within Solanaceae and, more generally, within plants [12].
Basically, to defend themselves, plants have developed a complex defense system to quickly recognize invading pathogens and transmit the message of attack [13,14]. The innate immunity system of plants has evolved in two recognition layers (PTI: PAMP (pathogen-associated molecular pattern)-triggered immunity and ETI: effector-triggered immunity). Plants’ own numerous non-self-recognition receptors are able to identify enemy molecules and induce a set of pathways and signaling cascades to repel attacks [15,16]. In ETI, immunity is mainly activated through the recognition of pathogen effectors via plant disease R-proteins. The major class of R-genes is represented by members of the nucleotide-binding site and leucine-rich repeat (NB-LRR or NLR) gene family [17]. The activation of NB-LRRs, during ETI response, induces programmed cell death, known as hypersensitive response (HR) [18,19]. Historically, NB-LRRs are divided into two subclasses, namely TIR-NB-LRR (TNL) and CC-NB-LRR (CNL) [20]. In addition to their role in pathogen recognition, some NB-LRR proteins contribute to signal transduction and/or amplification. Recent studies described a novel NB-LRR subclass, tagged as RPW8-NB-LRR (RNL, also known as helpers), whose protein members carry the RPW8 (resistance to powdery mildew 8) domain at the N terminal. RNLs mediate the immune response by interacting with NB-LRR “sensor” proteins involved in the detection of pathogens [21]. Additional NB-LRR helpers, which belong to a separate CNL subclass and exhibit high sequence similarity with NRC1 (NB-LRR protein required for HR-associated cell death 1), are required for cell death mediated by NB-LRR sensors [22]. NB-LRR helpers turned out to be essential in tomato, not only to support the activity of NB-LRR sensors, but also to counteract plant pathogens that evolve quickly, thus increasing the robustness of the innate immune system [23,24].
2. The Genome-Wide Arrangement of Tomato NB-LRR Genes
NB-LRR genes belong to a protein family with a very variable number of members among plants, ranging from about 50 to over 1000 [25]. A total of 294 NB-LRR genes were automatically identified and characterized by Andolfo et al. [5] and later revisited by Chandraprakash and Thomas [26] starting from the gene annotation released by the international Tomato Annotation Group (iTAG). However, that process failed to identify few genes, so the tomato NB-LRR complement was fully re-annotate by using the RenSeq method [3].
S. lycopersicum harbors approximately 320 NB-LRR-encoding genes arranged on all 12 chromosomes, whose genome-wide distribution is not random [3]. The largest number of NB-LRR genes is on chromosomes 4, 5, and 11 (~45%), whilst the smallest number is on chromosome 3 (9 genes), as found in other Solanaceae [12]. Some chromosomes predominantly host members of specific subclasses. Chromosomes 4 and 5 are particularly rich in CNLs. The largest number of TNLs (43%) is on chromosome 1, while chromosomes 3, 6, and 10 do not have them. Full-length RNL genes are only on chromosomes 2 and 4 [25], and are orthologous of the Arabidopsis thaliana NRG1 (N requirement gene 1) and Nicotiana benthamiana ADR1 (Activated Disease Resistance 1) R-gene helpers [27,28]; while tomato NRC1-homologs were located on chromosomes 2 and 10 [22].
The tomato NB-LRR repertoire includes also about 100 proteins that lack the full complement of domains that characterize genes within the NB-LRR subclasses. These include 14 CC-NB (CN) and three TIR-NBS (TN) proteins that have no LRR domain [3]. The majority of incomplete NB-LRR genes (~80%) only own a single domain and their function is still unknown, although it has been speculated they could act as adaptors or regulators of NB-LRR proteins with proven resistance activity [29]. In tomato the number of incomplete NB-LRR genes with evidence of expression is higher than observed in other plant species [3].
Generally, NB-LRR-encoding genes are clustered, as result of both segmental and tandem duplications [12,16]. Over 65% of tomato NB-LRRs are gathered in small genomic regions spanning 200 kb or less. One third of NB-LRRs (107 genes) are concentrated in 20 clusters [12]. This specific organization reflects genomic hotspots for diversification. The largest tomato cluster harbors 14 CNL genes in a region of ~110-kb in size on the short arm of chromosome 4. All members of this cluster share high sequence similarity with the wild potato derived R-genes R2, Rpi-blb3, and Rpi-abpt [30]. It is likely that functional R-genes, which have not yet been identified, are in this rapidly evolving cluster.
In tomato, several NB-LRR clusters comprised genes encoding proteins with high similarity to known and well-characterized R-genes [5]. Deciphering the evolutionary history of a gene cluster is essential not only to discover the functional specificity of each cluster-related allele, but also to provide insights into genome diversification by species. The increasing amount of cloned R-genes (Table 1) over the last two decades paved the way for the investigation of the evolutionary dynamics that generate novel resistance, which arise to match the changing patterns of pathogen virulence [31].
Table 1. List of cloned NB-LRR resistance genes in tomato and its wild relatives. Gene name, protein class, chromosome number and source of resistance for each R-gene were reported.
|
Gene Name |
Protein Class |
Chromosome |
Pathogen/Insect |
Source of Resistance |
Reference |
|---|---|---|---|---|---|
|
Bs4 |
TNL |
5 |
Xanthomonas campestris pv. vesicatoria |
S. lycopersicum |
Ballvora et al. [38] |
|
Hero |
CNL |
4 |
Globodera rostochiensis |
S. pimpinellifolium |
Ernst et al. [39] |
|
I-2 |
CNL |
11 |
Fusarium oxysporum f. sp. Lycopersici |
S. pimpinellifolium |
Simons et al. [37] |
|
Mi-1.2 |
CNL |
6 |
Melaydogyne spp.; Macrosiphum euphorbiae; Bemisia tabaci |
S. peruvianum |
Rossi et al. [36] Mahfouze et al. [40] |
|
Mi-9 |
CNL |
6 |
Meloidogyne spp. |
S. arcanum |
Jablonska et al. [41] |
|
Prf |
CNL |
5 |
Pseudomonas syringae |
S. pimpinellifolium |
Salmeron et al. [42] |
|
Ph-3 |
CNL |
9 |
Phitophthora infestans |
S. pimpinellifolium |
Zhang et al. [43] |
|
Sw-5 |
CNL |
9 |
Tomato spotted wilt virus |
S. peruvianum |
Brommonschenkel et al. [44] |
|
Tm-1 |
CNL |
2 |
Tomato mosaic virus |
S. habrochaites |
Ishibashi et al. [45] |
|
Tm-2 |
CNL |
9 |
Tobacco mosaic virus |
S. habrochaites |
Lanfermeijer et al. [46] |
|
Tm-2a |
CNL |
9 |
Tobacco mosaic virus |
S. peruvianum |
Lanfermeijer et al. [47] |
|
Ty 2 |
CNL |
11 |
Tomato leaf curly yellow virus |
S. habrochaites |
Yang et al. [48] |
Tomato NB-LRR loci are preferentially located in recombination hotspots, where meiotic crossovers are more frequent. Interestingly, all tomato cloned NB-LRR resistance genes, but not Tm22 and Prf conferring a fairly durable resistance [32,33], lie in regions exhibiting high/medium rates of recombination. The choice to retain the resistance loci into hot or cold recombination regions may reflect a different evolutionary state of pathogen–plant interactions. Recombination may be favorable in gene families controlling resistance to highly variable pathogens but unfavorable in families that control resistance to pathogens with low genetic plasticity [34]. The knowledge on the potential effects of crossover frequency on the tomato genome is prerequisite to predict R-gene haplotypes emerging following hybridization events.
The recombination density can increase the drift of genes in large clusters, such as I2 and Mi, which are in adaptive evolutionary state and have shown a frequency of recombination that is twice the average [35]. These two super-clusters, which include the R-genes Mi and I-2 [36,37], conferring resistance to Meloidogyne incognita and Fusarium oxysporum f. sp. Lycopersici respectively, have been extensively studied. The I2 super-cluster comprises seven NB-LRR genes gathered in a region of 390 kb on chromosome 11. The I-2 homolog genes were grouped in two sub-clusters of 54 kb and 28 kb in size. Similarly, six CNLs are in the Mi super-cluster, which is split into two sub-groups on chromosome 6. A region of approximately 400 kb was involved in the intra-chromosomal duplications and generated the Mi super-cluster [5]. In several plant genomes, the NB-LRR-encoding genes have been amplified, thus resulting in species-specific sub-families [16]. The diversification of tomato R-gene arsenal was mediated by duplication events of distinct NB-LRR paralogs (Figure 1). Forty-five out of ~320 NB-LRR sequences in tomato are more similar to each other than to any other non-Solanaceae sequences. Indeed, large gene expansions (more than 15 copies), involving unknown NB-LRRs—as well as Prf/R1, Hero/Mi 1.2, Gro1-4/N members—were observed.
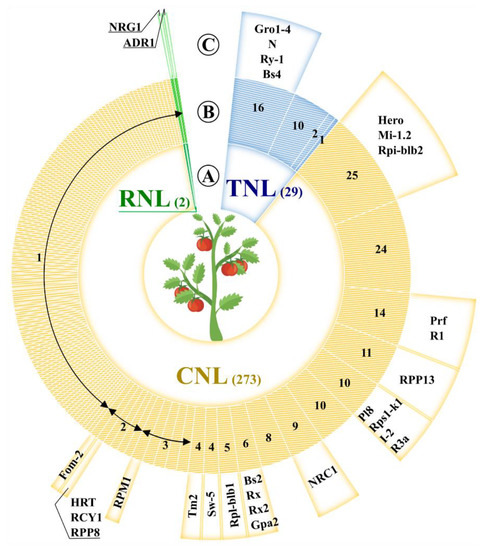
Figure 1. The tomato defense arsenal. (A) The full R-gene repertoire was displayed with respect to the NB-LRR subclasses (TNL in blue; CNL in orange and RNL in green). The total number of CNLs, TNLs, and RNLs was shown in brackets. (B) The NB-LRR paralogs identified by OrthoMCL with default settings were grouped (annular segments) and the amount of members in each group was specified. (C) For each group of paralogs, the well-characterized R-gene homolog was indicated (Table S1), when available.
3. Genomic-Driven Breeding for Developing New Resistant Tomato Varieties
The growing body of knowledge on the Solanaceae genomes will undoubtedly expedite the transfer of beneficial traits into tomato (Figure 2). A large number of R genes (including NB-LRR) have been introgressed from tomato wild relatives [64,65,66]. For example, sources of resistance against root-knot nematodes, aphids, whiteflies, viruses (TMV, TYLC, TSWV), and fungi were found in S. arcanum, S. habrochaites, S. pennelli and S. peruvianum, (Table 1). In addition, several genes derived for other Solanaceae species extended the tomato gene pool via transformation [67,68,69].
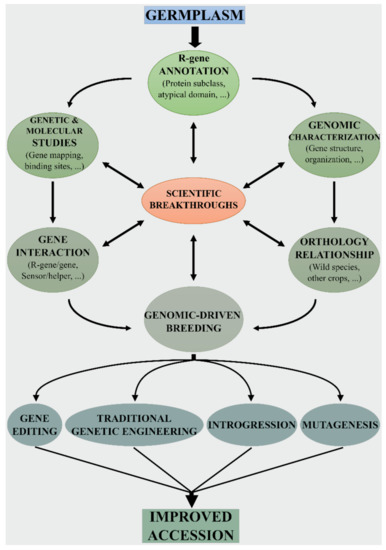
Traditional breeding based on “introgressomics” [70] greatly promoted the transfer of wild resistance genes in tomato. After the selection of the most eligible wild species and its hybridization with the cultivated species, the resistance traits are introgressed into the cultivated background after several generations of backcrossing. However, linkage drag (i.e., the undesirable effects of genes linked to the gene to be introgressed) is often associated with traditional introgression. As an example, the S. peruvianum introgression carrying the tomato mosaic virus (ToMV) resistance gene Tm22 can cover up to 79% of chromosome 9 in modern tomato varieties [71]. Therefore, it is highly desirable to establish reliable and more and more precise methods that can broaden genetic diversity through the introgression of alleles from wild relatives. The generation of new combinations of resistance alleles should be assisted by genomic-driven tomato breeding to minimize unwanted traits.
Genome editing technologies can make the “rewilding” process more easy [72], as they allow for the precise introduction of desirable genes/alleles from related wild relatives into elite cultivars (Figure 2). Indeed, plant disease resistance can be enhanced by targeting different genes of the plant defense machinery [73]. To edit NB-LRR genes it would be necessary modifying single nucleotides via “base-editing” and/or via homology-directed repair (HDR)-mediated base substitution [74]. In this case, homologous sequences serve as donors to repair site-specific double-strand DNA breaks caused by site-directed nucleases. Indeed, it is known that a few amino acid binding sites in different domains of NB-LRR proteins are required for pathogen recognition. Knowledge on the pathogen recognition sites of a particular NB-LRR receptor can be used to improve homologous NB-LRR in other accessions/species. As an example, Giannakopoulou et al. [75] assessed the degree of response of I2 random mutants to the Phytophthora infestans effector AVR3a. The mutant I2(I141N) conferred partial resistance to P. infestans and had a wider response spectrum to F. oxysporum f. sp. lycopersici effectors. Similarly, Segretin et al. [76] performed a gain-of-function random mutagenesis screen of the potato NB-LRR immune receptor R3a expanding its response to the P. infestans effector AVR3a. Remarkably, they found that the N336Y mutation conferred response also to the effector protein AVR3a4 from P. capsici.
To the best of our knowledge, however, no ‘base editing’ experiments or HDR-mediated base substitution has been attempted yet for NB-LRR genes in tomato or other Solanaceae. Genome editing technologies can also be used to edit or insert specific cis-regulatory elements into promoter regions, alter the epigenetic status [77] and to target miRNAs controlling the expression of NB-LRR genes with both cis- and trans-regulatory effects [78,79]. Indeed, it was observed that the silencing of miR482b in tomato plants promote the expression of several NBS–LRR receptors, thus enhancing resistance to P. infestans [80]. Transcriptional suppression may act as buffer for R-genes, thus reducing constraints on R-gene sequences [81].
Recent studies have shown that immune responses are mediated by complex and knotty networks of genes [23]. In some cases, the involvement of additional loci can make difficult the transfer of resistance traits in a new genetic background. For an optimal response to pathogens, it would therefore be appropriate to preserve almost entirely the wild NB-LRR network so that the new developed varieties will have greater ability to react to stresses. As a consequence, a viable and alternative solution for designing resistant tomato varieties is the de novo domestication of wild species [82,83]. With a particular focus on the concept of signaling by cooperative assembly formation, it would be desirable to exploit the wild genetic background by going to edit only key domestication/improvement genes [83,84,85]. Indeed, the expansion of the pathogen sensor functionality can require to bring together a large number of actors that can be activated in a proximity-based manner resulting in a higher-magnitude signal [86]. Understanding the evolution, assembly, and regulation of tomato immune receptor circuits is crucial to delivering fine-tuned tomato resistant varieties.
This entry is adapted from the peer-reviewed paper 10.3390/genes12020184