p53 is a 393-amino-acids-long transcription factor composed of a globular DNA binding domain flanked by a transcription activation domain in N-terminal and a tetramerization domain in the C-terminal part of the protein and is active in a homo-tetrameric state.
- p53
1. Introduction
The first mention of a “prion p53” actually dates back to 1995 [88]. As a basis to explain the dominant-negative effect of mutant p53 over the wild-type protein, Jo Milner and Elizabeth Medcalf evaluated the in vitro effect of several mutants of p53 (p. R151S, p. R247I, p. R273P, p. R273L) on wild-type p53. They showed that these mutants drive a wild-type p53 bearing a “wild-type” conformation [p53WT] toward a mutant conformation [p53MUT] when co-translated, thereby discriminating two in vitro allosteric variants of wild-type p53 [89].
The conformational flexibility of p53 has also provided grounds to the prion p53 hypothesis. Changes in p53 conformation have been monitored by their reactivity to conformation-specific antibodies [90]. For example, pAb1620 antibody recognizes the [p53WT] conformation while pAb240 [91] recognizes the [p53MUT] conformation. From there on, p53 mutants were classified as having wild-type [p53WT] or mutant [p53MUT] conformations based on their reactivity with either pAb1620 or pAb240 antibody. Mutants harboring a [p53WT] conformation were, however, not systematically transcriptionally active. These observations later gave rise to the subdivision of p53 mutants into two categories: (i)“DNA contact” mutants (R273H, R248Q, R248W), which have a decreased ability to bind DNA, and (ii) “conformation or structural” mutants (R175H, G245S, R249S, R282H), in which mutations induce a local to global destabilization of the protein structure [88,92] that exposes specific motives at the surface of the protein, thereby allowing reactivity with the pAb240 antibody. However, the relevance of these two classes of mutants is being challenged [93,94]. Some mutations such as p.V135A lead to a [p53WT] conformation when translated at 30 °C, but the protein undergoes a conversion to a [p53MUT] conformation within two minutes when the temperature shifts from 30 °C to 37 °C [95], also illustrating p53 conformational flexibility. Hence, the apparent ability of some p53 mutants to induce a conformational shift of a wild-type p53 protein bearing a [p53WT] conformation towards a [p53MUT] conformation, together with the existence of alternative conformations of p53 gave ground to the “prion p53” hypothesis [88,96], at a time when the prion protein PrPSc was in the limelight (Figure 2a).
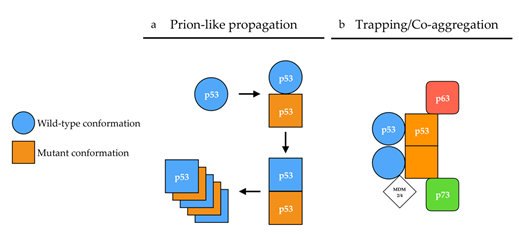
Figure 2. Proposed mechanisms of p53 aggregation. (a) According to the prion-like hypothesis, mutant p53 (orange) in a [p53MUT] conformation (square) induces a conformational shift of wild-type p53 (blue) from a [p53WT] (circle) conformation toward a [p53MUT] conformation in a prion-like manner. In a [p53MUT] conformation, wild-type and mutant p53 form amyloid fibers. (b) According to the co-aggregation/trapping hypothesis, mutant p53 hetero-tetramerizes with wild-type p53. The unfolded core of mutant p53 allows new interactions with p63/p73 isoforms and MDM2/4 in a trapping/co-aggregation mechanism.
2. p53 Aggregation in Tissue Samples and Tumor Cell Lines
p53 mutants have early on been described as forming aggregates in tissue samples and tumor cell lines. p53 has indeed been shown to form protein aggregates that abnormally accumulate in the nucleus and sometimes in the cytoplasm of cells from several types of tumor samples [97–102]. The aggregative features of p53 have been monitored for a time by immunohistochemistry (anti-p53 antibodies DO-1 and others) as a surrogate for the identification of tumors carrying p53 mutation [98,103–105], which is associated with worsened cancer prognosis (reviewed in [106]). However, immunohistochemistry often fails to deliver consistent results and is thus not the expected gold standard [28,104].
The amyloid nature of p53 aggregates has been mostly explored by co-localization experiments relying on anti-p53 antibodies, anti-fibrils antibodies (OC, A11 [107–109]) or amyloid dyes (thioflavin T, thioflavin S or Congo red). However, major variations are observed in the quantity and features of p53 aggregation, depending on the cell type, the mutation of p53 or the method used to quantify said aggregation [106,110–114], thereby rendering arduous any extensive comparison. In addition, the nature of the proteins forming the A11/OC-positive or amyloid dye-positive protein aggregates was not determined, thus the sometimes-partial co-localization of p53 with these amyloid aggregates precludes, for now, p53 from being classified as a genuine amyloid by the International Society of Amyloidosis nomenclature committee [36,115].
Studies concerning p53 aggregation have been limited to the canonical (full-length FL-p53ɑ) isoform of p53 and its mutant forms. However, recent reports focused on the potential aggregation of p53 isoforms ∆40p53 [116] and ∆133p53 [117,118] have opened new perspective to the field. The tissue-specific expression of p53 isoforms [15] could indeed partially account for the diversity of p53 aggregation phenotypes observed.
3. p53 Aggregation: In Vitro Dynamics
Despite the fact that only 36 proteins have been shown to form pathogenic amyloids in vivo, it is important to note that in vitro amyloid formation is observed for many more protein sequences. It is actually a property shared by many, if not all, natural polypeptide chains, once placed in the appropriate conditions [119–126], and it seems that p53 is no exception to the rule. Indeed, a large body of literature reports that, when subjected to high pressure, high temperature, zinc absence, low pH or RNA molecules, full-length p53, N-terminal, core or C-terminal fragments of p53, can be led to convert into either amorphous aggregates or ThT positive amyloid fibers in vitro [106,113,127–135].
The nucleation-dependent mechanism has three characteristics: the existence of a lag time, critical concentration and seeding. The lag time corresponds to the time required for nucleus formation during which the protein appears to be soluble. In contrast with nucleation-dependent polymerization, the growth of a linear polymer does not require nucleation and is characterized by the sequential buildup of intermediates. No lag time is observed, and supersaturated solutions rapidly aggregate. This process can be difficult to distinguish from a nucleation-dependent process with a very short lag time or from seeded growth [136]. The in vitro kinetics of p53 aggregation indeed differs from that of the classical nucleation-growth formation of amyloid fibrils, as the initiation of p53 aggregation happens to be relatively rapid [106,129,137] and as such is reminiscent of that of a linear polymer. The kinetics of p53 aggregation involves the formation of small aggregates that rapidly form amyloid structures that bind ThT and subsequently grow into larger amorphous aggregates. The progress curves fit to a two-step sequential pattern [138–140] in which a destabilized mutant p53 may co-aggregate with wild-type p53 and its paralogs p63 and p73. In this model, a mutant preferentially adopts an unfolded structure and would primarily react with another fast-unfolding mutant protein while only occasionally trapping a slow-unfolding wild-type protein. The mutant population rapidly self-aggregates before much of the wild-type p53 protein is depleted. However, as wild-type p53 is incorporated in hetero-tetramers by mutants, the continual production of mutant p53 in a cancer cell would gradually trap more and more wild-type p53 [141], its paralogs p63 and p73 [14,139] and MDM2 [142]. The trapping dynamics could also involve molecular chaperone HSP70 as it is proposed to stabilize mutant p.R175H and increase its aggregation [143]. This may account for the observations of co-aggregates in cell lines and in tumor tissue samples, and possibly causing a dominant-negative effect by directly impairing DNA binding activity [141,144,145].
4. p53 Aggregation: The Seeding Attempts
As nucleation is rate-limiting at low saturation levels, adding a seed (preformed nucleus) greatly accelerates the polymerization of molecules from solution [120,136,146–149]. However, in vitro p53 preformed aggregates did not significantly seed the aggregation of bulk proteins and stoichiometric amounts of aggregation-prone mutants induced only small amounts of wild-type p53 to co-aggregate [138–140]. p53 seeding experiments are indeed usually based on large amounts of aggregated proteins (10% of the dilution of aggregated proteins) [106] whereas very small amounts of prion or prionoid aggregates (1% to 0.05%) seed aggregation of bulk protein [147,150,151]. In 2013, based on prior demonstration of amyloid aggregation of p53 in vitro, Forget et al. tackled the question of whether or not an in vitro aggregated full-length p53 could be able to propagate its conformation to endogenous wild-type p53 in cells. These p53 aggregates were shown to penetrate cells via a nonspecific macropinocytosis pathway and to induce the co-aggregation of endogenous wild-type p53 in equally amorphous aggregates [152]. However, this report shows a single cell exhibiting p53 aggregates which hardly demonstrates that p53 can induce the aggregation of endogenous p53 proteins. Co-culture experiments were also used by Gosh et al. to demonstrate the cell-to-cell transmission of amyloid aggregates of p53 that were previously induced by the P8 peptide (PILTIITL, corresponding to the residues 250–257 of p53) [113]. The in vitro mechanism of p53 aggregation corresponds more to trapping by cross-reaction and co-aggregation rather than classical seeding and growth (see below; [139]).
5. p53 Aggregation: The Trapping Evidence
A consequence of prion self-propagation is that PrPC protein overexpression leads to an increased propagation of PrPSc prion due to the increased availability of its substrate [153,154]. This is also a feature of prionoids. However, the dominant-negative inhibition of mutants and isoforms of p53 has been shown to be dose-dependent in Soas-2 cells [155] as well as in S. cerevisiæ [14,156], two models lacking endogenous p53. Conversely, the expression of wild-type p53 has been shown to suppress the growth of tumor cell lines bearing dominant negative p53 mutants [157,158] and to overwhelm the inhibition exerted by mutant p53 on its transcription activity in baker’s yeast [14].
Then, in order to challenge the main ability of prions that is self-propagation, wild-type p53 has been co-expressed in yeast with a mutant p53 (p.R175H or p.R248Q) which expression has been placed under the control of a galactose-inducible promoter in a typical prion-propagation assay [159,160]. When co-expressed with mutant p53, wild-type p53 transcriptional functions were inhibited by the dominant-negative mutant p53. However, as soon as mutant p53 expression was shut off, wild-type p53 fully recovered its transcriptional abilities [14]. These results thus show that mutant p53 did not print its [p53MUT] conformation on wild-type p53 in a prion-like manner in S. cerevisiae, although recent data also suggest that when fused to EYFP, p53 overexpression leads, in some rare yeast cells, to the formation of aggregates that are transmitted across generations [161]. Altogether, these data show that the dominant-negative effect of mutant p53 is dose-dependent and can be reduced or even neutralized by increasing the wild-type/mutant p53 ratio.
The fact that increasing wild-type p53 expression level can overwhelm mutant p53 dominant-negative effect also led to p53-based gene therapy trials relying on adenoviruses. They aimed at restoring p53 tumor suppressive function in cancer cells by thwarting mutant p53 with a therapeutic wild-type p53 gene. Advexin and Gendicine [162] appeared as the main therapeutic candidates, having been used in numerous trials (including phase III). Chinese FDA approved the use of Gendicine in 2003 for head and neck tumors and in 2005 for naso-pharyngeal cancers [163]. To date, more than 30,000 patients have received Gendicine in association with chemo- or radiotherapy with promising results and relatively few side effects [164].
Other therapeutic strategies aim at reactivating mutant p53 tumor suppressor functions. Quinuclidines PRIMA-1 and its analog APR-246 (phase I and phase II clinical trials) have demonstrated their ability to inhibit cell proliferation and increase apoptosis in cancer cell lines and other models, although it has been reported that they could display non-p53 related activity [165]. The ReACp53 peptide targets the aggregation ability of mutant p53 and restores a partial function of the mutant protein, thereby inducing tumor shrinking both in vitro and in vivo [166,167]. These strategies, among others [168], seem to lead to mutant p53 refolding, allowing for a functional recovery of the protein.
Both the successful therapeutic approaches set up to target mutant p53 and the unsuccessful attempts of propagating [p53MUT] conformational phenotype strongly indicate that the p53 protein, be it in a wild-type or mutant state, does not behave as a prion nor as a prionoid.
This entry is adapted from the peer-reviewed paper 10.3390/cancers13020269