Skeletal muscle is composed of multinucleated, mature muscle cells (myofibers) responsible for contraction, and a resident pool of mononucleated muscle cell precursors (MCPs), that are maintained in a quiescent state in homeostatic conditions. Skeletal muscle is remarkable in its ability to adapt to mechanical constraints, a property referred as muscle plasticity and mediated by both MCPs and myofibers. This review summarizes recent insights into the mechanisms underlying nuclear force transmission in MCPs and myofibers.
- mechanotransduction
- Muscle disorders
- Nucleus
- nucleo-cytoplasmic coupling
- mechanics.
1. Introduction
2. Cytoskeletal Components Relevant for Force Transmission to the Nucleus
2.1 The perinuclear actin network and muscle differentiation
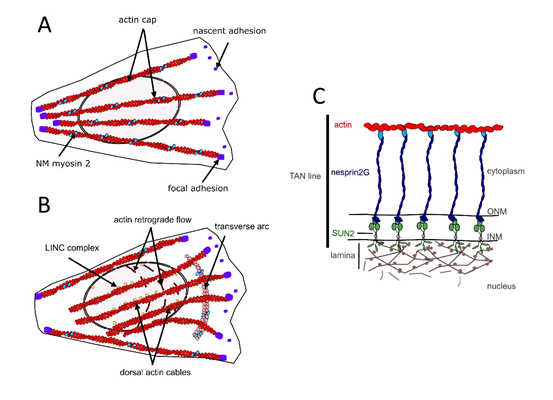
In different cell types, perinuclear actin emerges as a critical component for proper nucleo-cytoskeletal connections [39, 40, 46]. On the dorsal side of the nucleus of cells grown in 2D culture, perinuclear actin comprises the actin cap formed by dorsal stress fibers [47] (Fig. 2A) and the so-called transmembrane actin-associated nuclear (TAN) lines [48] (Fig. 2B,C). The actin cap is composed of thick parallel and highly contractile acto-myosin filaments, tightly connected to the nucleus and attached to basal focal adhesion sites on both extremities [47, 49-51]. The perinuclear actin cap accumulates upon mechanical stimulation [49, 50] and has important roles in nuclear mechanotransduction [50, 52].
Fig. 2. Components of the perinuclear actin network in MCPs. A) Actin cap formed by dorsal stress fibers. B) TAN lines C) Illustration of the molecular composition of a TAN line.
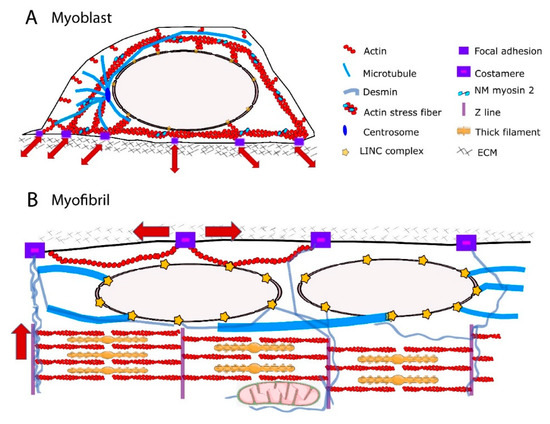
The actin cap is developmentally regulated, being present in myoblasts but absent in undifferentiated embryonic stem cells [53] and terminally differentiated muscle cells [27]. The structural and functional organization of actin cytoskeleton in the perinuclear region of myotubes remain partly unknown. During skeletal myofiber formation, nuclei are initially in the center of the myofiber and then move towards to myofiber periphery [54]. It has been shown that amphiphysin-2/BIN1, which is mutated in centronuclear myopathies, triggers peripheral nuclear positioning to the periphery of myofibers via N-WASP and actin, thus implicating the actin cytoskeleton in nuclear movement [55]. In addition, perinuclear actin may significantly alter the nuclear shape [27]. However, nuclear positioning to the myofiber periphery is mediated by centripetal forces arising from myofibril contraction around the nucleus [27]. Further, it has been proposed that a nucleus-cytoskeleton connection is not required for peripheral nuclear movement [27]. Future work should address how structural and functional connections between perinuclear actin network and nuclei are modified during skeletal myofiber formation. In addition to extensive cytoskeletal reorganization, shift in expression of actin components from non-muscle to muscle isoforms occur during skeletal myogenesis [56-58]. The muscle specific-isoform α-actin becomes the predominant actin in terminally differentiated myofibers and localizes to the sarcomeric thin filaments where it interacts with myosin to produce contractile force [59, 60]. The non-muscle actin g and β that are present around the nucleus in myoblasts [61] are downregulated during terminal differentiation of myoblasts into myotubes. In terminally differentiated myofibers, g- and β-actins reside in the cortical cytoskeleton and at costameres [62-65]. The costameric F-actin network is thought to contribute with other proteins to radial transmission of contractile force outward from the sarcomere to the extracellular matrix, adjacent muscle fibers and beyond [64]. Therefore, non-muscle F-actin could serve opposite force transmission direction according to the state of myogenic differentiation. The direction could be predominantly external to internal, toward NE in myoblasts, but predominantly internal and sarcomeric to external, toward extracellular matrix, in myofibers (Fig 1).
2.2 The MTs
MTs are three orders of magnitude stiffer than actin, IFs being the softness among the three major types of cytoskeleton filaments [65]. Their radial, centrosome-dominated distribution in myoblasts [66, 67] may favor the transmission of external mechanical forces to the NE and influence nuclear shape [68] and function [69] (Fig 1A). During the differentiation process, there is a large reorganization of the centrosome proteins: myoblasts possess a morphologically recognizable centrosome with characteristic marker proteins concentrated in the pericentriolar material, whereas myotube differentiation requires relocalization of centrosome proteins to the surface of the nucleus [67, 70, 71]. As centrosome proteins are critical for MT nucleation and/or anchoring, MT orientation is extensively redistributed into a more ordered paraxial array in myotubes [66, 67, 72, 73] (Fig 1B). Mature myofibers also exhibits a perinuclear network of MTs comprising a cage-like structure of a high-density meshwork that may be responsible for nuclear shaping and mechanical protection, and a circular and radial-anisotropic MTs, which are either polarized in the direction of contraction or in the lateral direction [74]. MT posttranslational modifications such as increased detyrosinated [75, 76] and binding of MTs to MT-associated proteins (MAPs), including EB1 and spectraplakin [74] confers stability to the MTs and have been shown to be essential for maintaining myonuclear morphology [74]. Additionally, it has been proposed that the spectrin domains of nesprin confers elastic features of the MT-spectraplakin-EB1 perinuclear network during contraction of striated muscle [74]. As a consequence, primary defects in the nuclear-associated networks of MTs have been implicated in strain-induced myonuclear damage [27, 74, 77].
2.3 Cytoplasmic IFs
IFs have emerged as a perfect candidate for maintaining proper nuclear mechano-response because they are able to resist high mechanical stresses, i.e. bending and stretching to a considerable degree [65]. IFs are surprisingly flexible [78-82] and can undergo strain-stiffening [83-85]. This is due to the short persistence length of intermediate filaments (1-3 μm) [65]. In the cytoplasm, they can form mechanically relevant links to each other, to other cytoskeletal filaments, to membrane complexes, and to internal organelles including the nucleus [82, 86] (Fig 1). These mechanical properties and interconnections enable the IFs to serve as mechanical stress absorbers that protects the cytoplasm and organelles, including the nucleus, against large deformations [51, 87, 88]. This idea is supported by the fact that IFs can withstand deformations of up to 300% of their initial length without rupturing [89]. Several IFs are expressed and developmentally regulated in human skeletal muscle cells [90-93]. Non muscle-specific proteins vimentin and nestin are expressed in MCPs and myoblasts and are downregulated during later differentiation [94]. Desmin, the muscle-specific IF protein, is expressed at low levels in MCPs and its expression continuously increases to become the prominent IF in mature myofibers [94, 95]. It can form copolymers with synemin, another non-muscle specific IF, around the α-actinin-rich Z-lines [92]. In undifferentiated myoblasts, vimentin and desmin are stably linked to the outer nuclear membrane [96] via plectin [97], thus contributing to the perinuclear cage-like structure. During terminal muscle differentiation, desmin accumulates and forms a three-dimensional network between the contractile apparatus, the extracellular matrix, and other cell organelles like mitochondria, T-tubules and nuclei [95, 98-100] (Fig. 1). Close to the nucleus, desmin filaments extend from the Z-lines of striated muscles towards the NE, where they interact with plectin. Terminal differentiation induced-desmin redistribution is associated with posttranslational modifications such as phosphorylation and ADP-ribosylation [101] which in turn regulate IF assembly and disassembly as well as interactions between IFs and other cell components and structures [102]. In mature muscle fibers, the primary role of desmin is to link adjacent myofibrils to each other and to the extracellular matrix, via costameres [39, 103-105]. Consequently, a functional reduction in desmin is associated with structural instability of the sarcomeres [106]. Accumulating evidence indicates that desmin is also crucial as a stress-transmitting and stress-signaling network [98, 107-110]. Desmin interactions with the nucleus are required to maintain nuclear architecture in cardiomyocytes [111] and to prevent nuclear and muscle damage in response to mechanical challenges [111, 112]. Future studies will determine the contribution of desmin scaffolds in myonucleus architecture and function.
- Mechanical linkages between the cytoskeleton and the nucleoskeleton
LINC complexes provide direct physical nucleo-cytoskeletal coupling between the cytoskeleton network and the NE [113, 114] (Fig. 3). The LINC complexes comprise outer nuclear transmembrane proteins, called nesprins (NE Spectrin-Repeat Proteins) defined by the Klarsicht-ANC1-Syne-homology (KASH) domain. This domain directly interacts with the luminal domain of the inner nuclear membrane proteins Sad1 and UNC-84 (SUN) proteins 1 (SUN1) or 2 (SUN2) [44, 113] within the perinuclear space of the nuclear envelope. SUN proteins form trimers and span the inner nuclear membrane, with their N-amino-terminal nucleoplasmic domains interacting with lamins and lamin associated proteins within the nucleoplasm [115]. By crossing the outer nuclear membrane, nesprins provide a mechanical link from the cytoskeleton to the nucleoskeleton.
To date, six genes encoding for different nesprins-1, -2, -3, -4 lymphoid-restricted membrane protein (LRMP) and KASH5 have been identified in mammals [97, 116, 117]. Giant nesprins-1 and -2 are ubiquitously expressed with highest representation in striated muscle [118, 119]. The SYNE-1 and SYNE-2 genes encode the nesprin-1G (1008 kD) and nesprin-2G (792 kD) respectively with calponin domains at their N-termini that bind the actin cytoskeleton [116]. Nesprins-1G and -2G also bind to the MT motors dynein and kinesin via their C-terminal cytoplasmic stretch [113, 120-122]. Kinesin-1 interacts with nesprin-1G and -2 via their LEWD motifs [119, 120].
SYNE-1 and SYNE-2 have multiple internal promotors giving rise to shorter nesprin isoforms which lack the actin-binding domain [119, 123] (Fig 3). Alternative splicing also generates short isoforms that lack the C-terminal KASH domain as well as short isoforms that lack both the KASH domain and CH domains [124].
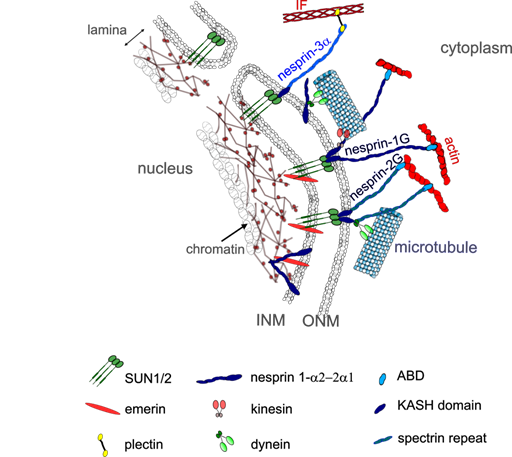
Fig. 3. LINC complexes in skeletal muscle. LINC is a complex of proteins including SUN1/2 and nesprins that connect the cytoskeleton to the nucleoskeleton. Different nesprin isoforms are expressed during myogenesis: in MCPs, the nesprin- 1G and -2G can interact with actin and microtubule in the cytoplasm and with SUN1/2 proteins, emerin and lamins on the inner nuclear membrane. Shorter nesprin-1α2 and nesprin-2α1 are expressed during myotube differentiation and can bind with microtubules in the cytoplasm via kinesin and other proteins such as A-kinase anchoring protein. Short nesprin-1a2 can also interact with intranuclear proteins such as lamins and emerin.
In contrast to SUN proteins, nesprins-1 and -2 switch localizations and isoforms during myogenesis [118, 119]. Nesprin-1 increases at the nuclear rim during early myogenesis but is partially replaced by nesprin-2 at later stages of muscle development [118, 119]. However, nesprin-1 appears to be critical in synaptic and non-synaptic myonuclear anchoring in skeletal muscle [125, 126], due to its ability to form interactions between myonuclei and actin cytoskeleton [125-127]. Expression of two shorter α isoforms, the nesprin-1α2 and nesprin-2α1 is switched on during myogenesis [128] [121,122] and becomes dominant in mature skeletal muscle [118]. They are found almost exclusively in skeletal and cardiac muscle [122, 128] and form a complex with emerin and A-type lamins at the inner nuclear membrane [129, 130]. At the outer nuclear membrane, nesprin-1α2 and nesprin-2α1 can interact with kinesin and microtubules [119, 123] (Fig. 3). Nesprin1-α2 is the main short form of nesprin-1 in skeletal muscle[131]. It is located mainly at the nuclear rim in early myotubes and immature muscle fibers, but then declines in most mature, adult muscle fibers [131], being restricted to neuromuscular junction nuclei [116, 119]. Nesprin1-α2 is required for correct positioning of myonuclei [77, 120, 132, 133] and MT nucleation from the NE [119], by recruiting A-Kinase Anchoring Protein-450 to the NE [77]. Nesprin-3 lacks actin-binding domains but can indirectly connect to the cytoskeleton by binding to another protein with tandem actin-binding calponin homology domain [134]. Though nesprin-3 exists as two isoforms, nesprin-3α and nesprin-3β, only nesprin-3α can attach to the cytoskeleton. For instance, nesprin-3α can anchor IFs to the NE through plectin [121-123, 126], a plakin family member that can also interact with actin filaments and MTs [97, 135-137]. This plectin-nesprin interaction requires the dimerization of plectin and takes place between the N-terminal actin-binding domain of plectin and the first spectrin repeat of nesprin-3α [135]. Nesprin-3β does not interact with Ifs because it lacks this spectrin-like repeat of nesprin-3α [135].
The different components of the LINC complexes have been associated with a number of pathogenic modifications in humans as well as in animal models. Perturbation of LINC complexes induces defective signal transduction across the NE [138, 139], prevented centrosome reorientation [48] and chromatin organization [77, 140-143] and abnormal nuclear positioning [116, 121, 131, 144-146]. It has been shown that mutations in nesprins-1 and -2 cause Emery-Dreifuss muscular dystrophy [77, 125, 147-150] and dilated cardiomyopathy [149]. It has been proposed that the giant nesprin-1 regulates a feedback loop by which MCPs adapt their intracellular tension to the softness of their native extracellular microenvironment through nucleo-cytoskeletal connections [150]. In addition, nesprin mutations can impair the interaction of nesprin with lamins, emerin and/or SUN proteins, thus affecting diverse functions including gene expression, nuclear shape and positioning [149]. As yet, no mutation in nesprin-3 has been found to be responsible for skeletal muscle diseases. However, acute depletion of nesprin-3 leads to rapid shrinkage and unfolding of nuclei in a microtubule-dependent manner in rat ventricular cardiomyocytes [111]. Loss of nuclear integrity is concomitant with compromised contractile function and has been proposed to contribute to the pathophysiological changes observed in desmin-related myopathies [111]. Further investigations are required to elucidate the complex mechanisms behind LINC-mediated nucleo-cytoskeletal linkages in skeletal muscle. Finally, although LINC complexes are critical for force transmission across the NE, alternative LINC-independent mechanisms have also been proposed [151]. For instance, it has been proposed that cell boundaries can drive nuclear flattening during cell spreading on rigid substrates [152]. It was shown that a direct compressive force by LINC anchored apical actin cables is not required for nuclear flattening [152]. According to this model, the overall nuclear shape is primarily dictated by passive forces generated within the actin cytoskeleton with cell spreading and forces transmitted by the actin cap or LINC complexes contribute to a lesser degree [151, 153].
- The nuclear lamina
The nuclear lamina is a filamentous network of proteins mainly composed of the type V IF lamin proteins that assemble into a meshwork underneath the inner nuclear membrane [154, 155]. The lamina is composed of lamins and lamin-associated proteins and provides structural support to the NE [156]. Lamins can be categorized as A-type (lamin A/C) or B-type (lamin B1, B2) lamins. They are key components of the nuclear environment and interact with a large number of proteins [140, 157-159], the nuclear membrane, and chromatin [157, 160] to influence mechanical cues and signaling pathways crucial for cellular proliferation and differentiation [161]. In addition, lamins are involved in the epigenetic regulation of chromatin with drastic consequences for gene regulation (review in [162]).
The B-type lamins, lamins B1 and B2, coded for by the LMNB1 and LMNB2 genes are expressed in all somatic cells. B-type lamins have an important role in nuclear shape [86, 163] and structure [155, 164, 165] and may provide nuclear elastic resistance [164] particularly in cells with low A-type lamins [86, 163, 166]. However, B-type lamin expression differs minimally across solid tissues or in response to matrix stiffness [167] and do not appear to play a major role in nuclear stiffness [86], In contrast, A-type lamins, encoded for by the LMNA gene, are critical for the appropriate nucleus stiffening [166] and dictate the nuclear strain stiffening that dominates nuclear resistance to large deformations [20]. Indeed, upon nuclear mechanostimulation, nucleoplasmic domain of the inner nuclear membrane protein emerin becomes phosphorylated by the protein proto-oncogene tyrosine protein kinase Sarcoma (Src) [168, 169]. The Ig fold domain of lamin A is able to partially unfold, leading to stretching of the protein [170]. A-type lamins undergoes dephosphorylation of the S22 residue, associated with relocalization of the nucleoplasmic fraction to the nuclear lamina [166, 168, 171]. This in turn reinforces the nuclear lamina by stabilization and assembly of A-type lamins and increases nuclear stiffness [161, 166]. Conversely, in reduced mechanical constraints, the mobility and turnover of A-type lamins increase [166, 171, 172]. It has also been shown that under compression, the coiled coils in the rod domains of A-type lamin polymers are able to slide over each other to contract the length of the rod, behaving as a compression spring able to absorb pressure [173]. The expression of A-type lamins can be correlated with tissue stiffness [166], stiff tissues such as muscle having higher A-type lamin expression and stiffer nuclei than those in softer tissues such as brain [166]. Moreover, the expression and stability of A-type lamins increase during myogenic differentiation [86], leading to nuclear stiffening [174].
Importantly, force-induced remodeling of the nuclear lamina may affect gene transcription by changing the binding properties of NE proteins and transcription factors. Indeed, it is known that chromatin containing actively transcribed genes exists in a less condensed state (i.e., euchromatin) compared to the more compact regions (i.e., heterochromatin) that contain silent genes. Chromatin contained in lamin-associated domains (LADs) is generally heterochromatin [175]. Force changes trigger rapid reorganization of the heterochromatin at the nuclear lamina and are associated with changes in global patterns of gene expression [176]. Nuclear stretch decreases the levels repressive histone H3K9me3 at the nuclear periphery and increases chromatin mobility [177]. According to recent studies from Wickström’ lab, this chromatin response relies on ER Ca2+ release [22]. A-type lamin levels and nuclear stiffness determine the sensitivity of the ER calcium release, where stiffer nuclei are more prone to respond [22]. Interestingly, myogenic differentiation is associated with specific developmental gene repositioning to and from the nuclear periphery, generally associated with repression of genes inhibitory to myogenesis and the activation of genes required for myotube differentiation [178]. Muscle-specific NE transmembrane proteins (NETs), including NET39, Transmembrane Protein 38A and wolframin ER transmembrane glycoprotein direct specific myogenic genes to the nuclear periphery to facilitate their repression and their combined knockdown almost completely blocks myotube formation [178]. There is also evidence that disrupted tethering of myogenic genes with NE [169,170] and muscle-specific NETs [178] could underlie muscle pathology in NE-linked diseases. Alternatively, NET-directed gene repositioning may contribute to nuclear stiffening during differentiation.
In line with these physiological roles of A-type lamins, mutations in the LMNA gene cause laminopathies, a heterogeneous group of disorders, including skeletal muscle dystrophies and cardiomyopathies [156, 179-182]. The severity of the muscle disease is highly variable, the most severe form being the LMNA-related congenital muscular dystrophy [183, 184]. Although the physiopathology of the disease still requires further studies, there is clear evidence that impaired integrity of the nucleus [184-188], aberrant positioning of myogenic genes [178, 189, 190] and defective mechanotransduction signaling [29, 31, 185, 191, 192] all contribute to the muscle diseases related to LMNA mutations. Future studies will precisely determine how the combination of mechanical uncoupling /epigenic factors and a signaling defect could drive these skeletal muscle disorders.
- Chromatin-mediated mechanoresponse
Whereas the lamina has been recognized for many years as a major contributor to nuclear stiffness, there is now evidence that chromatin and its histone modification state also contribute to nuclear mechanics independently of A-type lamins [20, 23, 193-196]. It has been proposed that chromatin dominates nuclear force responses at short extensions of < 30% strain [20]. Chromatin-based nuclear rigidity operates by inducing changes in histone modification state. Alterations that produce more euchromatin or heterochromatin result in decreased or increased small extension nuclear stiffness, respectively [20].
Upon mechanical stimulation, untethering LADs from the nuclear lamina could initiated gene repositioning and transcription. Mechanical forces could also decondense gene loci at the nuclear periphery, thus allowing better access for transcription machinery and increased transcription. However, it is important to remind that genes located at the nuclear envelope are not necessarily silent [197-199] and that untethering from the lamina is not sufficient to induce changes in gene transcription [200, 201]. Taken into account these limitations, there are evidence that force can induce chromatin rearrangement and gene activation. Indeed, the activation and transcription of many genes have been associated with effective force transmission to the nucleus and/or to nuclear deformations [184, 202-205]. In addition, force-induced chromatin reorganization could play a critical role in stem cell differentiation [178, 206, 207]. Interestingly, recent data show that forces propagate through lamina-chromatin interactions to directly stretch the chromatin and induce transcription upregulation in a living cell [208]. How the altered chromatin-mediated mechanoresponse contributes to mechanical load-mediated adaptation in normal and pathological skeletal muscle remains open for future studies.
- Nuclear positioning and mechanotransduction
Skeletal muscle fibers contain hundreds of flattened myonuclei evenly distributed at the periphery of each cell, with 3-8 nuclei (synaptic nuclei) anchored beneath the neuromuscular junction. How nuclei properly position themselves within each muscle fiber remains partly obscure especially in tissues. Myonuclear positioning in skeletal muscle cells is an active process that occurs during the differentiation and maturation process, as well as during regeneration [209]. It involves the cytoskeletal network of MTs, F-actin and/or IFs as follows : MTs in the initial translocation/spacing of nuclei along the fiber [54, 55, 77, 138, 210], and F-actin and desmin in their movement to the fiber periphery [27]. Mislocalization of myonuclei has been associated with a variety of muscle disorders characterized by muscle atrophy, muscle weakness and reduced muscle performance [209, 211].
The unique distribution of myonuclei at the muscle fiber periphery raises questions about the amount of intracellular force transmitted from the cytoskeleton to NE. Mispositioned myonuclei within individual multinucleated muscle fibers are a hallmark of many muscle diseases, including congenital myopathies and muscular dystrophies [55, 125, 138, 210, 212]. Abnormal nuclear positioning is likely to affect individual myonuclear activity by affecting force and strain transmission across the NE [74]. It has been proposed that centrally located myonuclei may experience higher contractile forces exerted by the myofibrils around the nucleus than peripheral nuclei which could disturb nuclear stability. However, whether or not mispositioned myonuclei are a cause or consequence of muscle disease states is currently still remains to be determined.
- Conclusions and future directions
An increasing number of studies focused on the importance of appropriate nuclear mechanotransduction for muscle homeostasis, regeneration and plasticity, have appeared in the literature over the last decade. Advances in deciphering the molecular mechanisms contributing to nuclear mechanotransduction strongly support the idea that defects in nuclear mechanotransduction contribute to human muscle disorders. Yet, an understanding of the mechanistic and physiological outcomes for nuclear mechanical stress response mainly arises from studies conducted in embryonic and/or mononucleated cells and may depend on the specific cell lines used. The majority of nuclear and cytoskeletal components involved in nuclear mechanotransduction are developmentally regulated and largely reorganized during muscle differentiation, which complicates the understanding of nuclear mechanotransduction defects in muscle disorders.
We anticipate that future research efforts will provide new insights into how the terminal differentiation of MCPs into multinucleated muscle fibers affect nuclear mechanotransduction. In addition, we foresee the elucidation of the contributive role of stress- and strain-induced nuclear response in normal and diseased striated muscles in the next decade.
Acknowledgments: The authors acknowledged the Association Institute of Myology, Paris France for support, USEK and Lebanon CNRS for SJ financial support, and Gillian Butler-Browne for careful reading.
Conflicts of Interest: Declare conflicts of interest or state “The authors declare no conflict of interest.”
References
.
- Abmayr, S.M.; Pavlath, G.K., Myoblast fusion: lessons from flies and mice. Development, 2012. 139(4): p. 641-656.
- Kirby, T.J.; Lammerding, J., Emerging views of the nucleus as a cellular mechanosensor. Nature Cell Biology, 2018. 20(4): p. 373-381.
- Ato, S.; Kido, K.; Sase, K.; Fujita, S., Response of Resistance Exercise-Induced Muscle Protein Synthesis and Skeletal Muscle Hypertrophy Are Not Enhanced After Disuse Muscle Atrophy in Rat. Frontiers in Physiology, 2020. 11: p. 469.
- Burkholder, T.J., Mechanotransduction in skeletal muscle. Frontiers in Bioscience: A Journal and Virtual Library, 2007. 12: p. 174-191.
- Masschelein, E.; D’Hulst, G.; Zvick, J.; Hinte, L.; Soro-Arnaiz, I.; Gorski, T.; von Meyenn, F.; Bar-Nur, O.; De Bock, K., Exercise promotes satellite cell contribution to myofibers in a load-dependent manner. Skeletal Muscle, 2020. 10(1): p. 21.
- Aureille, J.; Buffière‐Ribot, V.; Harvey, B.E.; Boyault, C.; Pernet, L.; Andersen, T.; Bacola, G.; Balland, M.; Fraboulet, S.; Van Landeghem, L.; Guilluy, C., Nuclear envelope deformation controls cell cycle progression in response to mechanical force. EMBO reports, 2019. 20(9).
- Uroz, M.; Wistorf, S.; Serra-Picamal, X.; Conte, V.; Sales-Pardo, M.; Roca-Cusachs, P.; Guimerà, R.; Trepat, X., Regulation of cell cycle progression by cell–cell and cell–matrix forces. Nature Cell Biology, 2018. 20(6): p. 646-654.
- Fukuda, S.; Kaneshige, A.; Kaji, T.; Noguchi, Y.-t.; Takemoto, Y.; Zhang, L.; Tsujikawa, K.; Kokubo, H.; Uezumi, A.; Maehara, K.; Harada, A.; Ohkawa, Y.; Fukada, S.-i., Sustained expression of HeyL is critical for the proliferation of muscle stem cells in overloaded muscle. eLife, 2019. 8: p. e48284.
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; Elvassore, N.; Piccolo, S., Role of YAP/TAZ in mechanotransduction. Nature, 2011. 474(7350): p. 179-183.
- Elosegui-Artola, A.; Andreu, I.; Beedle, A.E.M.; Lezamiz, A.; Uroz, M.; Kosmalska, A.J.; Oria, R.; Kechagia, J.Z.; Rico-Lastres, P.; Le Roux, A.-L.; Shanahan, C.M.; Trepat, X.; Navajas, D.; Garcia-Manyes, S.; Roca-Cusachs, P., Force Triggers YAP Nuclear Entry by Regulating Transport across Nuclear Pores. Cell, 2017. 171(6): p. 1397-1410.e14.
- Kumar, A.; Mazzanti, M.; Mistrik, M.; Kosar, M.; Beznoussenko, Galina V.; Mironov, Alexandre A.; Garrè, M.; Parazzoli, D.; Shivashankar, G.V.; Scita, G.; Bartek, J.; Foiani, M., ATR Mediates a Checkpoint at the Nuclear Envelope in Response to Mechanical Stress. Cell, 2014. 158(3): p. 633-646.
- Kidiyoor, G.R.; Li, Q.; Bastianello, G.; Bruhn, C.; Giovannetti, I.; Mohamood, A.; Beznoussenko, G.V.; Mironov, A.; Raab, M.; Piel, M.; Restuccia, U.; Matafora, V.; Bachi, A.; Barozzi, S.; Parazzoli, D.; Frittoli, E.; Palamidessi, A.; Panciera, T.; Piccolo, S.; Scita, G.; Maiuri, P.; Havas, K.M.; Zhou, Z.-W.; Kumar, A.; Bartek, J.; Wang, Z.-Q.; Foiani, M., ATR is essential for preservation of cell mechanics and nuclear integrity during interstitial migration. Nature Communications, 2020. 11(1): p. 4828.
- Itano, N.; Okamoto, S.-i.; Zhang, D.; Lipton, S.A.; Ruoslahti, E., Cell spreading controls endoplasmic and nuclear calcium: A physical gene regulation pathway from the cell surface to the nucleus. Proceedings of the National Academy of Sciences, 2003. 100(9): p. 5181-5186.
- Enyedi, B.; Jelcic, M.; Niethammer, P., The Cell Nucleus Serves as a Mechanotransducer of Tissue Damage-Induced Inflammation. Cell, 2016. 165(5): p. 1160-1170.
- Raab, M.; Gentili, M.; de Belly, H.; Thiam, H.-R.; Vargas, P.; Jimenez, A.J.; Lautenschlaeger, F.; Voituriez, R.; Lennon-Dumenil, A.-M.; Manel, N.; Piel, M., ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science, 2016. 352(6283): p. 359-362.
- Chen, N.Y.; Kim, P.H.; Fong, L.G.; Young, S.G., Nuclear membrane ruptures, cell death, and tissue damage in the setting of nuclear lamin deficiencies. Nucleus, 2020. 11(1): p. 237-249.
- Dahl, K.N.; Ribeiro, A.J.S.; Lammerding, J., Nuclear Shape, Mechanics, and Mechanotransduction. Circulation Research, 2008. 102(11): p. 1307-1318.
- Janota, C.S.; Calero-Cuenca, F.J.; Gomes, E.R., The role of the cell nucleus in mechanotransduction. Current Opinion in Cell Biology, 2020. 63: p. 204-211.
- Enyedi, B.; Niethammer, P., Nuclear membrane stretch and its role in mechanotransduction. Nucleus (Austin, Tex.), 2017. 8(2): p. 156-161.
- Stephens, A.D.; Banigan, E.J.; Adam, S.A.; Goldman, R.D.; Marko, J.F., Chromatin and lamin A determine two different mechanical response regimes of the cell nucleus. Molecular Biology of the Cell, 2017. 28(14): p. 1984-1996.
- Stephens, A.D.; Banigan, E.J.; Marko, J.F., Separate roles for chromatin and lamins in nuclear mechanics. Nucleus, 2018. 9(1): p. 119-124.
- Nava, M.M.; Miroshnikova, Y.A.; Biggs, L.C.; Whitefield, D.B.; Metge, F.; Boucas, J.; Vihinen, H.; Jokitalo, E.; Li, X.; García Arcos, J.M.; Hoffmann, B.; Merkel, R.; Niessen, C.M.; Dahl, K.N.; Wickström, S.A., Heterochromatin-Driven Nuclear Softening Protects the Genome against Mechanical Stress-Induced Damage. Cell, 2020. 181(4): p. 800-817.e22.
- Stephens, A.D.; Banigan, E.J.; Marko, J.F., Chromatin’s physical properties shape the nucleus and its functions. Current Opinion in Cell Biology, 2019. 58: p. 76-84.
- Flück, M.; Hoppeler, H., Molecular basis of skeletal muscle plasticity--from gene to form and function. Reviews of Physiology, Biochemistry and Pharmacology, 2003. 146: p. 159-216.
- Martino, F.; Perestrelo, A.R.; Vinarský, V.; Pagliari, S.; Forte, G., Cellular Mechanotransduction: From Tension to Function. Frontiers in Physiology, 2018. 9: p. 824.
- Essawy, N.; Samson, C.; Petitalot, A.; Moog, S.; Bigot, A.; Herrada, I.; Marcelot, A.; Arteni, A.-A.; Coirault, C.; Zinn-Justin, S., An Emerin LEM-Domain Mutation Impairs Cell Response to Mechanical Stress. Cells, 2019. 8(6): p. 570.
- Roman, W.; Martins, J.P.; Carvalho, F.A.; Voituriez, R.; Abella, J.V.G.; Santos, N.C.; Cadot, B.; Way, M.; Gomes, E.R., Myofibril contraction and crosslinking drive nuclear movement to the periphery of skeletal muscle. Nature Cell Biology, 2017. 19(10): p. 1189-1201.
- Fischer, M.; Rikeit, P.; Knaus, P.; Coirault, C., YAP-Mediated Mechanotransduction in Skeletal Muscle. Frontiers in Physiology, 2016. 7.
- Owens, D.J.; Fischer, M.; Jabre, S.; Moog, S.; Mamchaoui, K.; Butler-Browne, G.; Coirault, C., Lamin Mutations Cause Increased YAP Nuclear Entry in Muscle Stem Cells. Cells, 2020. 9(4).
- Jorgenson, K.W.; Phillips, S.M.; Hornberger, T.A., Identifying the Structural Adaptations that Drive the Mechanical Load-Induced Growth of Skeletal Muscle: A Scoping Review. Cells, 2020. 9(7): p. 1658.
- Owens, D.J.; Messeant, J.; Moog, S.; Viggars, M.; Ferry, A.; Mamchaoui, K.; Lacene, E.; Romero, N.; Brull, A.; Bonne, G.; Butler-Browne, G.; Coirault, C., Lamin-Related Congenital Muscular Dystrophy Alters Mechanical Signaling and Skeletal Muscle Growth. Int J Mol Sci, 2020. 22(1).
- D’Alessandro, M.; Hnia, K.; Gache, V.; Koch, C.; Gavriilidis, C.; Rodriguez, D.; Nicot, A.-S.; Romero, Norma B.; Schwab, Y.; Gomes, E.; Labouesse, M.; Laporte, J., Amphiphysin 2 Orchestrates Nucleus Positioning and Shape by Linking the Nuclear Envelope to the Actin and Microtubule Cytoskeleton. Developmental Cell, 2015. 35(2): p. 186-198.
- Kim, J.-K.; Louhghalam, A.; Lee, G.; Schafer, B.W.; Wirtz, D.; Kim, D.-H., Nuclear lamin A/C harnesses the perinuclear apical actin cables to protect nuclear morphology. Nature Communications, 2017. 8(1): p. 2123.
- Heo, S.-J.; Driscoll, T.P.; Thorpe, S.D.; Nerurkar, N.L.; Baker, B.M.; Yang, M.T.; Chen, C.S.; Lee, D.A.; Mauck, R.L., Differentiation alters stem cell nuclear architecture, mechanics, and mechano-sensitivity. eLife, 2016. 5: p. e18207.
- Onuh, J.O.; Qiu, H., Serum response factor-cofactor interactions and their implications in disease. The FEBS journal, 2020.
- Watt, K.I.; Goodman, C.A.; Hornberger, T.A.; Gregorevic, P., The Hippo Signaling Pathway in the Regulation of Skeletal Muscle Mass and Function. Exercise and Sport Sciences Reviews, 2018. 46(2): p. 92-96.
- Gnimassou, O.; Francaux, M.; Deldicque, L., Hippo Pathway and Skeletal Muscle Mass Regulation in Mammals: A Controversial Relationship. Frontiers in Physiology, 2017. 8: p. 190.
- Gabriel, B.M.; Hamilton, D.L.; Tremblay, A.M.; Wackerhage, H., The Hippo signal transduction network for exercise physiologists. Journal of Applied Physiology (Bethesda, Md.: 1985), 2016. 120(10): p. 1105-1117.
- Maniotis, A.J.; Chen, C.S.; Ingber, D.E., Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proceedings of the National Academy of Sciences, 1997. 94(3): p. 849-854.
- Wang, N.; Tytell, J.D.; Ingber, D.E., Mechanotransduction at a distance: mechanically coupling the extracellular matrix with the nucleus. Nature Reviews Molecular Cell Biology, 2009. 10(1): p. 75-82.
- Zhang, J.; Alisafaei, F.; Nikolić, M.; Nou, X.A.; Kim, H.; Shenoy, V.B.; Scarcelli, G., Nuclear Mechanics within Intact Cells Is Regulated by Cytoskeletal Network and Internal Nanostructures. Small, 2020. 16(18): p. 1907688.
- Ramdas, N.M.; Shivashankar, G.V., Cytoskeletal Control of Nuclear Morphology and Chromatin Organization. Journal of Molecular Biology, 2015. 427(3): p. 695-706.
- Haque, F.; Mazzeo, D.; Patel, J.T.; Smallwood, D.T.; Ellis, J.A.; Shanahan, C.M.; Shackleton, S., Mammalian SUN protein interaction networks at the inner nuclear membrane and their role in laminopathy disease processes. The Journal of Biological Chemistry, 2010. 285(5): p. 3487-3498.
- Crisp, M.; Burke, B., The nuclear envelope as an integrator of nuclear and cytoplasmic architecture. FEBS letters, 2008. 582(14): p. 2023-2032.
- Crisp, M.; Liu, Q.; Roux, K.; Rattner, J.B.; Shanahan, C.; Burke, B.; Stahl, P.D.; Hodzic, D., Coupling of the nucleus and cytoplasm: role of the LINC complex. The Journal of Cell Biology, 2006. 172(1): p. 41-53.
- Ingber, D.E., Cellular mechanotransduction: putting all the pieces together again. The FASEB Journal, 2006. 20(7): p. 811-827.
- Khatau, S.B.; Hale, C.M.; Stewart-Hutchinson, P.J.; Patel, M.S.; Stewart, C.L.; Searson, P.C.; Hodzic, D.; Wirtz, D., A perinuclear actin cap regulates nuclear shape. Proceedings of the National Academy of Sciences, 2009. 106(45): p. 19017-19022.
- Luxton, G.W.G.; Gomes, E.R.; Folker, E.S.; Vintinner, E.; Gundersen, G.G., Linear arrays of nuclear envelope proteins harness retrograde actin flow for nuclear movement. Science (New York, N.Y.), 2010. 329(5994): p. 956-959.
- Kim, D.-H.; Chambliss, A.B.; Wirtz, D., The multi-faceted role of the actin cap in cellular mechanosensation and mechanotransduction. Soft Matter, 2013. 9(23): p. 5516.
- Chambliss, A.B.; Khatau, S.B.; Erdenberger, N.; Robinson, D.K.; Hodzic, D.; Longmore, G.D.; Wirtz, D., The LINC-anchored actin cap connects the extracellular milieu to the nucleus for ultrafast mechanotransduction. Scientific Reports, 2013. 3(1): p. 1087.
- Neelam, S.; Chancellor, T.J.; Li, Y.; Nickerson, J.A.; Roux, K.J.; Dickinson, R.B.; Lele, T.P., Direct force probe reveals the mechanics of nuclear homeostasis in the mammalian cell. Proceedings of the National Academy of Sciences, 2015. 112(18): p. 5720-5725.
- Shiu, J.-Y.; Aires, L.; Lin, Z.; Vogel, V., Nanopillar force measurements reveal actin-cap-mediated YAP mechanotransduction. Nature Cell Biology, 2018. 20(3): p. 262-271.
- Khatau, S.B.; Kusuma, S.; Hanjaya-Putra, D.; Mali, P.; Cheng, L.; Lee, J.S.H.; Gerecht, S.; Wirtz, D., The Differential Formation of the LINC-Mediated Perinuclear Actin Cap in Pluripotent and Somatic Cells. PLoS ONE, 2012. 7(5): p. e36689.
- Cadot, B.; Gache, V.; Gomes, E.R., Moving and positioning the nucleus in skeletal muscle - one step at a time. Nucleus (Austin, Tex.), 2015. 6(5): p. 373-381.
- Falcone, S.; Roman, W.; Hnia, K.; Gache, V.; Didier, N.; Lainé, J.; Auradé, F.; Marty, I.; Nishino, I.; Charlet‐Berguerand, N.; Romero, N.B.; Marazzi, G.; Sassoon, D.; Laporte, J.; Gomes, E.R., N‐ WASP is required for Amphiphysin‐2/ BIN 1‐dependent nuclear positioning and triad organization in skeletal muscle and is involved in the pathophysiology of centronuclear myopathy. EMBO Molecular Medicine, 2014. 6(11): p. 1455-1475.
- Lloyd, C.M.; Berendse, M.; Lloyd, D.G.; Schevzov, G.; Grounds, M.D., A novel role for non-muscle γ-actin in skeletal muscle sarcomere assembly. Experimental Cell Research, 2004. 297(1): p. 82-96.
- Sanger, J.W.; Kang, S.; Siebrands, C.C.; Freeman, N.; Du, A.; Wang, J.; Stout, A.L.; Sanger, J.M., How to build a myofibril. Journal of Muscle Research and Cell Motility, 2006. 26(6-8): p. 343-354.
- Hotulainen, P.; Lappalainen, P., Stress fibers are generated by two distinct actin assembly mechanisms in motile cells. Journal of Cell Biology, 2006. 173(3): p. 383-394.
- Bains, W.; Ponte, P.; Blau, H.; Kedes, L., Cardiac actin is the major actin gene product in skeletal muscle cell differentiation in vitro. Molecular and Cellular Biology, 1984. 4(8): p. 1449-1453.
- Lin, J.J.; Lin, J.L., Assembly of different isoforms of actin and tropomyosin into the skeletal tropomyosin-enriched microfilaments during differentiation of muscle cells in vitro. The Journal of Cell Biology, 1986. 103(6): p. 2173-2183.
- Otey, C.A.; Kalnoski, M.H.; Bulinski, J.C., Immunolocalization of muscle and nonmuscle isoforms of actin in myogenic cells and adult skeletal muscle. Cell Motility and the Cytoskeleton, 1988. 9(4): p. 337-348.
- Craig, S.W.; Pardo, J.V., Gamma actin, spectrin, and intermediate filament proteins colocalize with vinculin at costameres, myofibril-to-sarcolemma attachment sites. Cell Motility, 1983. 3(5): p. 449-462.
- Rybakova, I.N.; Patel, J.R.; Ervasti, J.M., The Dystrophin Complex Forms a Mechanically Strong Link between the Sarcolemma and Costameric Actin. Journal of Cell Biology, 2000. 150(5): p. 1209-1214.
- Ervasti, J.M., Costameres: the Achilles' Heel of Herculean Muscle. Journal of Biological Chemistry, 2003. 278(16): p. 13591-13594.
- Pegoraro, A.F.; Janmey, P.; Weitz, D.A., Mechanical Properties of the Cytoskeleton and Cells. Cold Spring Harbor Perspectives in Biology, 2017. 9(11): p. a022038.
- Musa, H.; Orton, C.; Morrison, E.E.; Peckham, M., Microtubule assembly in cultured myoblasts and myotubes following nocodazole induced microtubule depolymerisation. J Muscle Res Cell Motil, 2003. 24(4-6): p. 301-8.
- Becker, R.; Leone, M.; Engel, F., Microtubule Organization in Striated Muscle Cells. Cells, 2020. 9(6): p. 1395.
- Chang, W.; Worman, H.J.; Gundersen, G.G., Accessorizing and anchoring the LINC complex for multifunctionality. Journal of Cell Biology, 2015. 208(1): p. 11-22.
- Webster, M.; Witkin, K.L.; Cohen-Fix, O., Sizing up the nucleus: nuclear shape, size and nuclear-envelope assembly. Journal of Cell Science, 2009. 122(10): p. 1477-1486.
- Starr, D.A., Muscle Development: Nucleating Microtubules at the Nuclear Envelope. Current Biology, 2017. 27(19): p. R1071-R1073.
- Srsen, V.; Fant, X.; Heald, R.; Rabouille, C.; Merdes, A., Centrosome proteins form an insoluble perinuclear matrix during muscle cell differentiation. BMC Cell Biology, 2009. 10(1): p. 28.
- Warren, R.H., MICROTUBULAR ORGANIZATION IN ELONGATING MYOGENIC CELLS. The Journal of Cell Biology, 1974. 63(2): p. 550-566.
- Pizon, V.; Gerbal, F.; Diaz, C.C.; Karsenti, E., Microtubule-dependent transport and organization of sarcomeric myosin during skeletal muscle differentiation. The EMBO Journal, 2005. 24(21): p. 3781-3792.
- Wang, S.; Reuveny, A.; Volk, T., Nesprin provides elastic properties to muscle nuclei by cooperating with spectraplakin and EB1. Journal of Cell Biology, 2015. 209(4): p. 529-538.
- Mian, I.; Pierre-Louis, W.S.; Dole, N.; Gilberti, R.M.; Dodge-Kafka, K.; Tirnauer, J.S., LKB1 Destabilizes Microtubules in Myoblasts and Contributes to Myoblast Differentiation. PLoS ONE, 2012. 7(2): p. e31583.
- Gundersen, G.G.; Khawaja, S.; Bulinski, J.C., Generation of a stable, posttranslationally modified microtubule array is an early event in myogenic differentiation. The Journal of Cell Biology, 1989. 109(5): p. 2275-2288.
- Gimpel, P.; Lee, Y.L.; Sobota, R.M.; Calvi, A.; Koullourou, V.; Patel, R.; Mamchaoui, K.; Nédélec, F.; Shackleton, S.; Schmoranzer, J.; Burke, B.; Cadot, B.; Gomes, E.R., Nesprin-1α-Dependent Microtubule Nucleation from the Nuclear Envelope via Akap450 Is Necessary for Nuclear Positioning in Muscle Cells. Current Biology, 2017. 27(19): p. 2999-3009.e9.
- Block, J.; Schroeder, V.; Pawelzyk, P.; Willenbacher, N.; Köster, S., Physical properties of cytoplasmic intermediate filaments. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, 2015. 1853(11): p. 3053-3064.
- Fudge, D.S.; Gardner, K.H.; Forsyth, V.T.; Riekel, C.; Gosline, J.M., The mechanical properties of hydrated intermediate filaments: insights from hagfish slime threads. Biophysical Journal, 2003. 85(3): p. 2015-2027.
- Kreplak, L.; Bär, H.; Leterrier, J.F.; Herrmann, H.; Aebi, U., Exploring the mechanical behavior of single intermediate filaments. Journal of Molecular Biology, 2005. 354(3): p. 569-577.
- Wagner, O.I.; Rammensee, S.; Korde, N.; Wen, Q.; Leterrier, J.-F.; Janmey, P.A., Softness, strength and self-repair in intermediate filament networks. Experimental Cell Research, 2007. 313(10): p. 2228-2235.
- Kim, S.; Coulombe, P.A., Intermediate filament scaffolds fulfill mechanical, organizational, and signaling functions in the cytoplasm. Genes & Development, 2007. 21(13): p. 1581-1597.
- Block, J.; Witt, H.; Candelli, A.; Peterman, E.J.G.; Wuite, G.J.L.; Janshoff, A.; Köster, S., Nonlinear Loading-Rate-Dependent Force Response of Individual Vimentin Intermediate Filaments to Applied Strain. Physical Review Letters, 2017. 118(4): p. 048101.
- Lorenz, C.; Forsting, J.; Schepers, A.V.; Kraxner, J.; Bauch, S.; Witt, H.; Klumpp, S.; Köster, S., Lateral Subunit Coupling Determines Intermediate Filament Mechanics. Physical Review Letters, 2019. 123(18): p. 188102.
- Smoler, M.; Coceano, G.; Testa, I.; Bruno, L.; Levi, V., Apparent stiffness of vimentin intermediate filaments in living cells and its relation with other cytoskeletal polymers. Biochim Biophys Acta Mol Cell Res, 2020. 1867(8): p. 118726.
- Lammerding, J.; Fong, L.G.; Ji, J.Y.; Reue, K.; Stewart, C.L.; Young, S.G.; Lee, R.T., Lamins A and C but Not Lamin B1 Regulate Nuclear Mechanics. Journal of Biological Chemistry, 2006. 281(35): p. 25768-25780.
- Patteson, A.E.; Vahabikashi, A.; Pogoda, K.; Adam, S.A.; Mandal, K.; Kittisopikul, M.; Sivagurunathan, S.; Goldman, A.; Goldman, R.D.; Janmey, P.A., Vimentin protects cells against nuclear rupture and DNA damage during migration. Journal of Cell Biology, 2019. 218(12): p. 4079-4092.
- Hu, J.; Li, Y.; Hao, Y.; Zheng, T.; Gupta, S.K.; Parada, G.A.; Wu, H.; Lin, S.; Wang, S.; Zhao, X.; Goldman, R.D.; Cai, S.; Guo, M., High stretchability, strength, and toughness of living cells enabled by hyperelastic vimentin intermediate filaments. Proceedings of the National Academy of Sciences, 2019. 116(35): p. 17175-17180.
- Kreplak, L.; Herrmann, H.; Aebi, U., Tensile properties of single desmin intermediate filaments. Biophys J, 2008. 94(7): p. 2790-9.
- Banwell, B.L., Intermediate filament-related myopathies. Pediatric Neurology, 2001. 24(4): p. 257-263.
- Paulin, D.; Huet, A.; Khanamyrian, L.; Xue, Z., Desminopathies in muscle disease: Desminopathies in muscle disease. The Journal of Pathology, 2004. 204(4): p. 418-427.
- Paulin, D.; Hovhannisyan, Y.; Kasakyan, S.; Agbulut, O.; Li, Z.; Xue, Z., Synemin-related skeletal and cardiac myopathies: an overview of pathogenic variants. American Journal of Physiology-Cell Physiology, 2020. 318(4): p. C709-C718.
- Capetanaki, Y.; Bloch, R.J.; Kouloumenta, A.; Mavroidis, M.; Psarras, S., Muscle intermediate filaments and their links to membranes and membranous organelles. Experimental Cell Research, 2007. 313(10): p. 2063-2076.
- Sejersen, T.; Lendahl, U., Transient expression of the intermediate filament nestin during skeletal muscle development. Journal of Cell Science, 1993. 106 ( Pt 4): p. 1291-1300.
- Lazarides, E.; Hubbard, B.D., Immunological characterization of the subunit of the 100 A filaments from muscle cells. Proceedings of the National Academy of Sciences, 1976. 73(12): p. 4344-4348.
- Mermelstein, C.S.; Andrade, L.R.; Portilho, D.M.; Costa, M.L., Desmin filaments are stably associated with the outer nuclear surface in chick myoblasts. Cell and Tissue Research, 2006. 323(2): p. 351-357.
- Wilhelmsen, K.; Litjens, S.H.; Kuikman, I.; Tshimbalanga, N.; Janssen, H.; van den Bout, I.; Raymond, K.; Sonnenberg, A., Nesprin-3, a novel outer nuclear membrane protein, associates with the cytoskeletal linker protein plectin. J Cell Biol, 2005. 171(5): p. 799-810.
- Lazarides, E., Intermediate filaments as mechanical integrators of cellular space. Nature, 1980. 283(5744): p. 249-256.
- Capetanaki, Y., Desmin Cytoskeleton A Potential Regulator of Muscle Mitochondrial Behavior and Function. Trends in Cardiovascular Medicine, 2002. 12(8): p. 339-348.
- Reipert, S., Association of Mitochondria with Plectin and Desmin Intermediate Filaments in Striated Muscle. Experimental Cell Research, 1999. 252(2): p. 479-491.
- Winter, D.L.; Paulin, D.; Mericskay, M.; Li, Z., Posttranslational modifications of desmin and their implication in biological processes and pathologies. Histochemistry and Cell Biology, 2014. 141(1): p. 1-16.
- Snider, N.T.; Omary, M.B., Post-translational modifications of intermediate filament proteins: mechanisms and functions. Nature Reviews Molecular Cell Biology, 2014. 15(3): p. 163-177.
- Gao, Q.Q.; McNally, E.M., The Dystrophin Complex: Structure, Function, and Implications for Therapy, in Comprehensive Physiology, R. Terjung, Editor. 2015, John Wiley & Sons, Inc.: Hoboken, NJ, USA. p. 1223-1239.
- Boudriau, S.; Vincent, M.; Côté, C.H.; Rogers, P.A., Cytoskeletal structure of skeletal muscle: identification of an intricate exosarcomeric microtubule lattice in slow- and fast-twitch muscle fibers. Journal of Histochemistry & Cytochemistry, 1993. 41(7): p. 1013-1021.
- Wang, K.; Ramirez-Mitchell, R., A network of transverse and longitudinal intermediate filaments is associated with sarcomeres of adult vertebrate skeletal muscle. The Journal of Cell Biology, 1983. 96(2): p. 562-570.
- Li, Z.; Colucci-Guyon, E.; Pinçon-Raymond, M.; Mericskay, M.; Pournin, S.; Paulin, D.; Babinet, C., Cardiovascular Lesions and Skeletal Myopathy in Mice Lacking Desmin. Developmental Biology, 1996. 175(2): p. 362-366.
- Price, M.G., Molecular analysis of intermediate filament cytoskeleton--a putative load-bearing structure. The American Journal of Physiology, 1984. 246(4 Pt 2): p. H566-572.
- Galou, M.; Gao, J.; Humbert, J.; Mericskay, M.; Li, Z.; Paulin, D.; Vicart, P., The importance of intermediate filaments in the adaptation of tissues to mechanical stress: evidence from gene knockout studies. Biology of the Cell, 1997. 89(2): p. 85-97.
- Tolstonog, G.V.; Sabasch, M.; Traub, P., Cytoplasmic Intermediate Filaments Are Stably Associated with Nuclear Matrices and Potentially Modulate Their DNA-Binding Function. DNA and Cell Biology, 2002. 21(3): p. 213-239.
- Boriek, A.M.; Capetanaki, Y.; Hwang, W.; Officer, T.; Badshah, M.; Rodarte, J.; Tidball, J.G., Desmin integrates the three-dimensional mechanical properties of muscles. American Journal of Physiology-Cell Physiology, 2001. 280(1): p. C46-C52.
- Heffler, J.; Shah, P.P.; Robison, P.; Phyo, S.; Veliz, K.; Uchida, K.; Bogush, A.; Rhoades, J.; Jain, R.; Prosser, B.L., A Balance Between Intermediate Filaments and Microtubules Maintains Nuclear Architecture in the Cardiomyocyte. Circulation Research, 2020. 126(3).
- Langer, H.T.; Mossakowski, A.A.; Willis, B.J.; Grimsrud, K.N.; Wood, J.A.; Lloyd, K.C.K.; Zbinden-Foncea, H.; Baar, K., Generation of desminopathy in rats using CRISPR-Cas9: Generation of desminopathy in rats using CRISPR-Cas9. Journal of Cachexia, Sarcopenia and Muscle, 2020.
- Starr, D.A.; Fridolfsson, H.N., Interactions Between Nuclei and the Cytoskeleton Are Mediated by SUN-KASH Nuclear-Envelope Bridges. Annual Review of Cell and Developmental Biology, 2010. 26(1): p. 421-444.
- Lombardi, M.L.; Lammerding, J., Keeping the LINC: the importance of nucleocytoskeletal coupling in intracellular force transmission and cellular function. Biochemical Society Transactions, 2011. 39(6): p. 1729-1734.
- Torbati, M.; Lele, T.P.; Agrawal, A., An Unresolved LINC in the Nuclear Envelope. Cellular and Molecular Bioengineering, 2016. 9(2): p. 252-257.
- Zhang, Q.; Skepper, J.N.; Yang, F.; Davies, J.D.; Hegyi, L.; Roberts, R.G.; Weissberg, P.L.; Ellis, J.A.; Shanahan, C.M., Nesprins: a novel family of spectrin-repeat-containing proteins that localize to the nuclear membrane in multiple tissues. Journal of Cell Science, 2001. 114(Pt 24): p. 4485-4498.
- Roux, K.J.; Crisp, M.L.; Liu, Q.; Kim, D.; Kozlov, S.; Stewart, C.L.; Burke, B., Nesprin 4 is an outer nuclear membrane protein that can induce kinesin-mediated cell polarization. Proceedings of the National Academy of Sciences of the United States of America, 2009. 106(7): p. 2194-2199.
- Randles, K.N.; Lam, L.T.; Sewry, C.A.; Puckelwartz, M.; Furling, D.; Wehnert, M.; McNally, E.M.; Morris, G.E., Nesprins, but not sun proteins, switch isoforms at the nuclear envelope during muscle development. Developmental Dynamics, 2010. 239(3): p. 998-1009.
- Holt, I.; Fuller, H.R.; Lam, L.T.; Sewry, C.A.; Shirran, S.L.; Zhang, Q.; Shanahan, C.M.; Morris, G.E., Nesprin-1-alpha2 associates with kinesin at myotube outer nuclear membranes, but is restricted to neuromuscular junction nuclei in adult muscle. Scientific Reports, 2019. 9(1): p. 14202.
- Wilson, M.H.; Holzbaur, E.L.F., Nesprins anchor kinesin-1 motors to the nucleus to drive nuclear distribution in muscle cells. Development, 2015. 142(1): p. 218-228.
- Chapman, M.A.; Zhang, J.; Banerjee, I.; Guo, L.T.; Zhang, Z.; Shelton, G.D.; Ouyang, K.; Lieber, R.L.; Chen, J., Disruption of both nesprin 1 and desmin results in nuclear anchorage defects and fibrosis in skeletal muscle. Human Molecular Genetics, 2014. 23(22): p. 5879-5892.
- Zhang, Q., Nesprin-2 is a multi-isomeric protein that binds lamin and emerin at the nuclear envelope and forms a subcellular network in skeletal muscle. Journal of Cell Science, 2005. 118(4): p. 673-687.
- Tapley, E.C.; Starr, D.A., Connecting the nucleus to the cytoskeleton by SUN–KASH bridges across the nuclear envelope. Current Opinion in Cell Biology, 2013. 25(1): p. 57-62.
- Rajgor, D.; Mellad, J.A.; Autore, F.; Zhang, Q.; Shanahan, C.M., Multiple Novel Nesprin-1 and Nesprin-2 Variants Act as Versatile Tissue-Specific Intracellular Scaffolds. PLoS ONE, 2012. 7(7): p. e40098.
- Puckelwartz, M.J.; Kessler, E.; Zhang, Y.; Hodzic, D.; Randles, K.N.; Morris, G.; Earley, J.U.; Hadhazy, M.; Holaska, J.M.; Mewborn, S.K.; Pytel, P.; McNally, E.M., Disruption of nesprin-1 produces an Emery Dreifuss muscular dystrophy-like phenotype in mice. Human Molecular Genetics, 2009. 18(4): p. 607-620.
- Zhang, X.; Xu, R.; Zhu, B.; Yang, X.; Ding, X.; Duan, S.; Xu, T.; Zhuang, Y.; Han, M., Syne-1 and Syne-2 play crucial roles in myonuclear anchorage and motor neuron innervation. Development, 2007. 134(5): p. 901-908.
- Zhang, J.; Felder, A.; Liu, Y.; Guo, L.T.; Lange, S.; Dalton, N.D.; Gu, Y.; Peterson, K.L.; Mizisin, A.P.; Shelton, G.D.; Lieber, R.L.; Chen, J., Nesprin 1 is critical for nuclear positioning and anchorage. Human Molecular Genetics, 2010. 19(2): p. 329-341.
- Duong, N.T.; Morris, G.E.; Lam, L.T.; Zhang, Q.; Sewry, C.A.; Shanahan, C.M.; Holt, I., Nesprins: Tissue-Specific Expression of Epsilon and Other Short Isoforms. PLoS ONE, 2014. 9(4): p. e94380.
- Mislow, J.M.K.; Holaska, J.M.; Kim, M.S.; Lee, K.K.; Segura-Totten, M.; Wilson, K.L.; McNally, E.M., Nesprin-1α self-associates and binds directly to emerin and lamin A in vitro. FEBS Letters, 2002. 525(1-3): p. 135-140.
- Wheeler, M.A.; Davies, J.D.; Zhang, Q.; Emerson, L.J.; Hunt, J.; Shanahan, C.M.; Ellis, J.A., Distinct functional domains in nesprin-1α and nesprin-2β bind directly to emerin and both interactions are disrupted in X-linked Emery–Dreifuss muscular dystrophy. Experimental Cell Research, 2007. 313(13): p. 2845-2857.
- Holt, I.; Duong, N.T.; Zhang, Q.; Lam, L.T.; Sewry, C.A.; Mamchaoui, K.; Shanahan, C.M.; Morris, G.E., Specific localization of nesprin-1-α2, the short isoform of nesprin-1 with a KASH domain, in developing, fetal and regenerating muscle, using a new monoclonal antibody. BMC Cell Biology, 2016. 17(1): p. 26.
- Roman, W.; Gomes, E.R., Nuclear positioning in skeletal muscle. Seminars in Cell & Developmental Biology, 2018. 82: p. 51-56.
- Zhou, C.; Rao, L.; Shanahan, C.M.; Zhang, Q., Nesprin-1/2: roles in nuclear envelope organisation, myogenesis and muscle disease. Biochemical Society Transactions, 2018. 46(2): p. 311-320.
- Liao, L.; Qu, R.; Ouang, J.; Dai, J., A Glance at the Nuclear Envelope Spectrin Repeat Protein 3. BioMed Research International, 2019. 2019: p. 1-8.
- Ketema, M.; Wilhelmsen, K.; Kuikman, I.; Janssen, H.; Hodzic, D.; Sonnenberg, A., Requirements for the localization of nesprin-3 at the nuclear envelope and its interaction with plectin. Journal of Cell Science, 2007. 120(19): p. 3384-3394.
- Wiche, G., Role of plectin in cytoskeleton organization and dynamics. Journal of Cell Science, 1998. 111 ( Pt 17): p. 2477-2486.
- Staszewska, I.; Fischer, I.; Wiche, G., Plectin isoform 1-dependent nuclear docking of desmin networks affects myonuclear architecture and expression of mechanotransducers. Human Molecular Genetics, 2015. 24(25): p. 7373-7389.
- Folker, E.S.; Östlund, C.; Luxton, G.W.G.; Worman, H.J.; Gundersen, G.G., Lamin A variants that cause striated muscle disease are defective in anchoring transmembrane actin-associated nuclear lines for nuclear movement. Proceedings of the National Academy of Sciences, 2011. 108(1): p. 131-136.
- Ho, C.Y.; Lammerding, J., Lamins at a glance. Journal of Cell Science, 2012. 125(Pt 9): p. 2087-2093.
- Hieda, M., Signal Transduction across the Nuclear Envelope: Role of the LINC Complex in Bidirectional Signaling. Cells, 2019. 8(2): p. 124.
- Burke, B.; Roux, K.J., Nuclei Take a Position: Managing Nuclear Location. Developmental Cell, 2009. 17(5): p. 587-597.
- Fridolfsson, H.N.; Ly, N.; Meyerzon, M.; Starr, D.A., UNC-83 coordinates kinesin-1 and dynein activities at the nuclear envelope during nuclear migration. Developmental Biology, 2010. 338(2): p. 237-250.
- Jain, N.; Iyer, K.V.; Kumar, A.; Shivashankar, G.V., Cell geometric constraints induce modular gene-expression patterns via redistribution of HDAC3 regulated by actomyosin contractility. Proceedings of the National Academy of Sciences, 2013. 110(28): p. 11349-11354.
- Lei, K.; Zhang, X.; Ding, X.; Guo, X.; Chen, M.; Zhu, B.; Xu, T.; Zhuang, Y.; Xu, R.; Han, M., SUN1 and SUN2 play critical but partially redundant roles in anchoring nuclei in skeletal muscle cells in mice. Proceedings of the National Academy of Sciences, 2009. 106(25): p. 10207-10212.
- Wu, Y.K.; Umeshima, H.; Kurisu, J.; Kengaku, M., Nesprins and opposing microtubule motors generate a point force that drives directional nuclear motion in migrating neurons. Development, 2018. 145(5): p. dev158782.
- Kengaku, M., Cytoskeletal control of nuclear migration in neurons and non-neuronal cells. Proceedings of the Japan Academy, Series B, 2018. 94(9): p. 337-349.
- Zhang, Q.; Bethmann, C.; Worth, N.F.; Davies, J.D.; Wasner, C.; Feuer, A.; Ragnauth, C.D.; Yi, Q.; Mellad, J.A.; Warren, D.T.; Wheeler, M.A.; Ellis, J.A.; Skepper, J.N.; Vorgerd, M.; Schlotter-Weigel, B.; Weissberg, P.L.; Roberts, R.G.; Wehnert, M.; Shanahan, C.M., Nesprin-1 and -2 are involved in the pathogenesis of Emery–Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Human Molecular Genetics, 2007. 16(23): p. 2816-2833.
- Stroud, M.J.; Feng, W.; Zhang, J.; Veevers, J.; Fang, X.; Gerace, L.; Chen, J., Nesprin 1α2 is essential for mouse postnatal viability and nuclear positioning in skeletal muscle. Journal of Cell Biology, 2017. 216(7): p. 1915-1924.
- Zhou, C.; Li, C.; Zhou, B.; Sun, H.; Koullourou, V.; Holt, I.; Puckelwartz, M.J.; Warren, D.T.; Hayward, R.; Lin, Z.; Zhang, L.; Morris, G.E.; McNally, E.M.; Shackleton, S.; Rao, L.; Shanahan, C.M.; Zhang, Q., Novel nesprin-1 mutations associated with dilated cardiomyopathy cause nuclear envelope disruption and defects in myogenesis. Human Molecular Genetics, 2017. 26(12): p. 2258-2276.
- Schwartz, C.; Fischer, M.; Mamchaoui, K.; Bigot, A.; Lok, T.; Verdier, C.; Duperray, A.; Michel, R.; Holt, I.; Voit, T.; Quijano-Roy, S.; Bonne, G.; Coirault, C., Lamins and nesprin-1 mediate inside-out mechanical coupling in muscle cell precursors through FHOD1. Scientific Reports, 2017. 7(1): p. 1253.
- Jahed, Z.; Mofrad, M.R., The nucleus feels the force, LINCed in or not! Current Opinion in Cell Biology, 2019. 58: p. 114-119.
- Li, Y.; Lovett, D.; Zhang, Q.; Neelam, S.; Kuchibhotla, Ram A.; Zhu, R.; Gundersen, Gregg G.; Lele, Tanmay P.; Dickinson, Richard B., Moving Cell Boundaries Drive Nuclear Shaping during Cell Spreading. Biophysical Journal, 2015. 109(4): p. 670-686.
- Szczesny, S.E.; Mauck, R.L., The Nuclear Option: Evidence Implicating the Cell Nucleus in Mechanotransduction. Journal of Biomechanical Engineering, 2017. 139(2): p. 021006.
- Aebi, U.; Cohn, J.; Buhle, L.; Gerace, L., The nuclear lamina is a meshwork of intermediate-type filaments. Nature, 1986. 323(6088): p. 560-564.
- Turgay, Y.; Eibauer, M.; Goldman, A.E.; Shimi, T.; Khayat, M.; Ben-Harush, K.; Dubrovsky-Gaupp, A.; Sapra, K.T.; Goldman, R.D.; Medalia, O., The molecular architecture of lamins in somatic cells. Nature, 2017. 543(7644): p. 261-264.
- Burke, B.; Stewart, C.L., The nuclear lamins: flexibility in function. Nature Reviews Molecular Cell Biology, 2013. 14(1): p. 13-24.
- Simon, D.N.; Wilson, K.L., The nucleoskeleton as a genome-associated dynamic 'network of networks'. Nature Reviews Molecular Cell Biology, 2011. 12(11): p. 695-708.
- Korfali, N.; Wilkie, G.S.; Swanson, S.K.; Srsen, V.; de las Heras, J.; Batrakou, D.G.; Malik, P.; Zuleger, N.; Kerr, A.R.W.; Florens, L.; Schirmer, E.C., The nuclear envelope proteome differs notably between tissues. Nucleus, 2012. 3(6): p. 552-564.
- Hieda, M., Implications for Diverse Functions of the LINC Complexes Based on the Structure. Cells, 2017. 6(1).
- Kalinowski, A.; Qin, Z.; Coffey, K.; Kodali, R.; Buehler, Markus J.; Lösche, M.; Dahl, Kris N., Calcium Causes a Conformational Change in Lamin A Tail Domain that Promotes Farnesyl-Mediated Membrane Association. Biophysical Journal, 2013. 104(10): p. 2246-2253.
- Osmanagic-Myers, S.; Dechat, T.; Foisner, R., Lamins at the crossroads of mechanosignaling. Genes & Development, 2015. 29(3): p. 225-237.
- Bianchi, A.; Manti, P.G.; Lucini, F.; Lanzuolo, C., Mechanotransduction, nuclear architecture and epigenetics in Emery Dreifuss Muscular Dystrophy: tous pour un, un pour tous. Nucleus, 2018. 9(1): p. 321-335.
- Coffinier, C.; Jung, H.J.; Nobumori, C.; Chang, S.; Tu, Y.; Barnes, R.H., 2nd; Yoshinaga, Y.; de Jong, P.J.; Vergnes, L.; Reue, K.; Fong, L.G.; Young, S.G., Deficiencies in lamin B1 and lamin B2 cause neurodevelopmental defects and distinct nuclear shape abnormalities in neurons. Mol Biol Cell, 2011. 22(23): p. 4683-93.
- Dahl, K.N., The nuclear envelope lamina network has elasticity and a compressibility limit suggestive of a molecular shock absorber. Journal of Cell Science, 2004. 117(20): p. 4779-4786.
- Shimi, T.; Kittisopikul, M.; Tran, J.; Goldman, A.E.; Adam, S.A.; Zheng, Y.; Jaqaman, K.; Goldman, R.D., Structural organization of nuclear lamins A, C, B1, and B2 revealed by superresolution microscopy. Molecular Biology of the Cell, 2015. 26(22): p. 4075-4086.
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.D.P.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.-W.; Tewari, M.; Rehfeldt, F.; Speicher, D.W.; Discher, D.E., Nuclear Lamin-A Scales with Tissue Stiffness and Enhances Matrix-Directed Differentiation. Science, 2013. 341(6149): p. 1240104-1240104.
- Buxboim, A.; Irianto, J.; Swift, J.; Athirasala, A.; Shin, J.-W.; Rehfeldt, F.; Discher, D.E., Coordinated increase of nuclear tension and lamin-A with matrix stiffness outcompetes lamin-B receptor that favors soft tissue phenotypes. Molecular Biology of the Cell, 2017. 28(23): p. 3333-3348.
- Guilluy, C.; Burridge, K., Nuclear mechanotransduction: Forcing the nucleus to respond. Nucleus, 2015. 6(1): p. 19-22.
- Guilluy, C.; Osborne, L.D.; Van Landeghem, L.; Sharek, L.; Superfine, R.; Garcia-Mata, R.; Burridge, K., Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nature Cell Biology, 2014. 16(4): p. 376-381.
- Bera, M.; Kotamarthi, H.C.; Dutta, S.; Ray, A.; Ghosh, S.; Bhattacharyya, D.; Ainavarapu, S.R.K.; Sengupta, K., Characterization of Unfolding Mechanism of Human Lamin A Ig Fold by Single-Molecule Force Spectroscopy—Implications in EDMD. Biochemistry, 2014. 53(46): p. 7247-7258.
- Buxboim, A.; Swift, J.; Irianto, J.; Spinler, Kyle R.; Dingal, P.C.Dave P.; Athirasala, A.; Kao, Y.-Ruei C.; Cho, S.; Harada, T.; Shin, J.-W.; Discher, Dennis E., Matrix Elasticity Regulates Lamin-A,C Phosphorylation and Turnover with Feedback to Actomyosin. Current Biology, 2014. 24(16): p. 1909-1917.
- Xia, Y.; Pfeifer, C.R.; Cho, S.; Discher, D.E.; Irianto, J., Nuclear mechanosensing. Emerging Topics in Life Sciences, 2018. 2(5): p. 713-725.
- Makarov, A.A.; Zou, J.; Houston, D.R.; Spanos, C.; Solovyova, A.S.; Cardenal-Peralta, C.; Rappsilber, J.; Schirmer, E.C., Lamin A molecular compression and sliding as mechanisms behind nucleoskeleton elasticity. Nature Communications, 2019. 10(1): p. 3056.
- Pajerowski, J.D.; Dahl, K.N.; Zhong, F.L.; Sammak, P.J.; Discher, D.E., Physical plasticity of the nucleus in stem cell differentiation. Proceedings of the National Academy of Sciences, 2007. 104(40): p. 15619-15624.
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; van Steensel, B., Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature, 2008. 453(7197): p. 948-51.
- Miroshnikova, Y.A.; Nava, M.M.; Wickström, S.A., Emerging roles of mechanical forces in chromatin regulation. Journal of Cell Science, 2017. 130(14): p. 2243-2250.
- Le, H.Q.; Ghatak, S.; Yeung, C.-Y.C.; Tellkamp, F.; Günschmann, C.; Dieterich, C.; Yeroslaviz, A.; Habermann, B.; Pombo, A.; Niessen, C.M.; Wickström, S.A., Mechanical regulation of transcription controls Polycomb-mediated gene silencing during lineage commitment. Nature Cell Biology, 2016. 18(8): p. 864-875.
- Robson, Michael I.; de las Heras, Jose I.; Czapiewski, R.; Lê Thành, P.; Booth, Daniel G.; Kelly, David A.; Webb, S.; Kerr, Alastair R.W.; Schirmer, Eric C., Tissue-Specific Gene Repositioning by Muscle Nuclear Membrane Proteins Enhances Repression of Critical Developmental Genes during Myogenesis. Molecular Cell, 2016. 62(6): p. 834-847.
- Donnaloja, F.; Carnevali, F.; Jacchetti, E.; Raimondi, M.T., Lamin A/C Mechanotransduction in Laminopathies. Cells, 2020. 9(5): p. 1306.
- Gruenbaum, Y.; Foisner, R., Lamins: Nuclear Intermediate Filament Proteins with Fundamental Functions in Nuclear Mechanics and Genome Regulation. Annual Review of Biochemistry, 2015. 84(1): p. 131-164.
- Janin, A.; Gache, V., Nesprins and Lamins in Health and Diseases of Cardiac and Skeletal Muscles. Frontiers in Physiology, 2018. 9: p. 1277.
- Brull, A.; Morales Rodriguez, B.; Bonne, G.; Muchir, A.; Bertrand, A.T., The Pathogenesis and Therapies of Striated Muscle Laminopathies. Frontiers in Physiology, 2018. 9: p. 1533.
- Quijano-Roy, S.; Mbieleu, B.; Bönnemann, C.G.; Jeannet, P.-Y.; Colomer, J.; Clarke, N.F.; Cuisset, J.-M.; Roper, H.; De Meirleir, L.; D'Amico, A.; Ben Yaou, R.; Nascimento, A.; Barois, A.; Demay, L.; Bertini, E.; Ferreiro, A.; Sewry, C.A.; Romero, N.B.; Ryan, M.; Muntoni, F.; Guicheney, P.; Richard, P.; Bonne, G.; Estournet, B., De novo LMNA mutations cause a new form of congenital muscular dystrophy. Annals of Neurology, 2008. 64(2): p. 177-186.
- Lammerding, J.; Schulze, P.C.; Takahashi, T.; Kozlov, S.; Sullivan, T.; Kamm, R.D.; Stewart, C.L.; Lee, R.T., Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. The Journal of Clinical Investigation, 2004. 113(3): p. 370-378.
- Lammerding, J.; Hsiao, J.; Schulze, P.C.; Kozlov, S.; Stewart, C.L.; Lee, R.T., Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. The Journal of Cell Biology, 2005. 170(5): p. 781-791.
- Hale, C.M.; Shrestha, A.L.; Khatau, S.B.; Stewart-Hutchinson, P.J.; Hernandez, L.; Stewart, C.L.; Hodzic, D.; Wirtz, D., Dysfunctional connections between the nucleus and the actin and microtubule networks in laminopathic models. Biophysical Journal, 2008. 95(11): p. 5462-5475.
- Zhang, Q.; Tamashunas, A.C.; Agrawal, A.; Torbati, M.; Katiyar, A.; Dickinson, R.B.; Lammerding, J.; Lele, T.P., Local, transient tensile stress on the nuclear membrane causes membrane rupture. Molecular Biology of the Cell, 2019. 30(7): p. 899-906.
- Earle, A.J.; Kirby, T.J.; Fedorchak, G.R.; Isermann, P.; Patel, J.; Iruvanti, S.; Moore, S.A.; Bonne, G.; Wallrath, L.L.; Lammerding, J., Mutant lamins cause nuclear envelope rupture and DNA damage in skeletal muscle cells. Nature Materials, 2020. 19(4): p. 464-473.
- Solovei, I.; Wang, Audrey S.; Thanisch, K.; Schmidt, Christine S.; Krebs, S.; Zwerger, M.; Cohen, Tatiana V.; Devys, D.; Foisner, R.; Peichl, L.; Herrmann, H.; Blum, H.; Engelkamp, D.; Stewart, Colin L.; Leonhardt, H.; Joffe, B., LBR and Lamin A/C Sequentially Tether Peripheral Heterochromatin and Inversely Regulate Differentiation. Cell, 2013. 152(3): p. 584-598.
- Mattout, A.; Pike, Brietta L.; Towbin, Benjamin D.; Bank, Erin M.; Gonzalez-Sandoval, A.; Stadler, Michael B.; Meister, P.; Gruenbaum, Y.; Gasser, Susan M., An EDMD Mutation in C. elegans Lamin Blocks Muscle-Specific Gene Relocation and Compromises Muscle Integrity. Current Biology, 2011. 21(19): p. 1603-1614.
- Emerson, L.J.; Holt, M.R.; Wheeler, M.A.; Wehnert, M.; Parsons, M.; Ellis, J.A., Defects in cell spreading and ERK1/2 activation in fibroblasts with lamin A/C mutations. Biochimica Et Biophysica Acta, 2009. 1792(8): p. 810-821.
- Bertrand, A.T.; Ziaei, S.; Ehret, C.; Duchemin, H.; Mamchaoui, K.; Bigot, A.; Mayer, M.; Quijano-Roy, S.; Desguerre, I.; Lainé, J.; Ben Yaou, R.; Bonne, G.; Coirault, C., Cellular microenvironments reveal defective mechanosensing responses and elevated YAP signaling in LMNA-mutated muscle precursors. Journal of Cell Science, 2014. 127(Pt 13): p. 2873-2884.
- Furusawa, T.; Rochman, M.; Taher, L.; Dimitriadis, E.K.; Nagashima, K.; Anderson, S.; Bustin, M., Chromatin decompaction by the nucleosomal binding protein HMGN5 impairs nuclear sturdiness. Nature Communications, 2015. 6: p. 6138.
- Krause, M.; Te Riet, J.; Wolf, K., Probing the compressibility of tumor cell nuclei by combined atomic force-confocal microscopy. Physical Biology, 2013. 10(6): p. 065002.
- Lherbette, M.; Dos Santos, Á.; Hari-Gupta, Y.; Fili, N.; Toseland, C.P.; Schaap, I.A.T., Atomic Force Microscopy micro-rheology reveals large structural inhomogeneities in single cell-nuclei. Scientific Reports, 2017. 7(1): p. 8116.
- Schäpe, J.; Prausse, S.; Radmacher, M.; Stick, R., Influence of lamin A on the mechanical properties of amphibian oocyte nuclei measured by atomic force microscopy. Biophysical Journal, 2009. 96(10): p. 4319-4325.
- Hubner, M.R.; Spector, D.L., Chromatin dynamics. Annu Rev Biophys, 2010. 39: p. 471-89.
- Sexton, T.; Schober, H.; Fraser, P.; Gasser, S.M., Gene regulation through nuclear organization. Nat Struct Mol Biol, 2007. 14(11): p. 1049-55.
- Jost, K.L.; Rottach, A.; Milden, M.; Bertulat, B.; Becker, A.; Wolf, P.; Sandoval, J.; Petazzi, P.; Huertas, D.; Esteller, M.; Kremmer, E.; Leonhardt, H.; Cardoso, M.C., Generation and characterization of rat and mouse monoclonal antibodies specific for MeCP2 and their use in X-inactivation studies. PLoS One, 2011. 6(11): p. e26499.
- Peric-Hupkes, D.; van Steensel, B., Role of the Nuclear Lamina in Genome Organization and Gene Expression. Cold Spring Harbor Symposia on Quantitative Biology, 2010. 75(0): p. 517-524.
- Schubeler, D.; Francastel, C.; Cimbora, D.M.; Reik, A.; Martin, D.I.; Groudine, M., Nuclear localization and histone acetylation: a pathway for chromatin opening and transcriptional activation of the human beta-globin locus. Genes Dev, 2000. 14(8): p. 940-50.
- Thomas, C.H.; Collier, J.H.; Sfeir, C.S.; Healy, K.E., Engineering gene expression and protein synthesis by modulation of nuclear shape. Proc Natl Acad Sci U S A, 2002. 99(4): p. 1972-7.
- Heo, S.J.; Nerurkar, N.L.; Baker, B.M.; Shin, J.W.; Elliott, D.M.; Mauck, R.L., Fiber stretch and reorientation modulates mesenchymal stem cell morphology and fibrous gene expression on oriented nanofibrous microenvironments. Ann Biomed Eng, 2011. 39(11): p. 2780-90.
- Driscoll, T.P.; Cosgrove, B.D.; Heo, S.J.; Shurden, Z.E.; Mauck, R.L., Cytoskeletal to Nuclear Strain Transfer Regulates YAP Signaling in Mesenchymal Stem Cells. Biophys J, 2015. 108(12): p. 2783-93.
- Banerjee, I.; Zhang, J.; Moore-Morris, T.; Pfeiffer, E.; Buchholz, K.S.; Liu, A.; Ouyang, K.; Stroud, M.J.; Gerace, L.; Evans, S.M.; McCulloch, A.; Chen, J., Targeted ablation of nesprin 1 and nesprin 2 from murine myocardium results in cardiomyopathy, altered nuclear morphology and inhibition of the biomechanical gene response. PLoS Genet, 2014. 10(2): p. e1004114.
- Martins, R.P.; Finan, J.D.; Guilak, F.; Lee, D.A., Mechanical regulation of nuclear structure and function. Annu Rev Biomed Eng, 2012. 14: p. 431-55.
- Jakkaraju, S.; Zhe, X.; Pan, D.; Choudhury, R.; Schuger, L., TIPs are tension-responsive proteins involved in myogenic versus adipogenic differentiation. Developmental Cell, 2005. 9(1): p. 39-49.
- Tajik, A.; Zhang, Y.; Wei, F.; Sun, J.; Jia, Q.; Zhou, W.; Singh, R.; Khanna, N.; Belmont, A.S.; Wang, N., Transcription upregulation via force-induced direct stretching of chromatin. Nature Materials, 2016. 15(12): p. 1287-1296.
- Folker, E.S.; Baylies, M.K., Nuclear positioning in muscle development and disease. Frontiers in Physiology, 2013. 4.
- Metzger, T.; Gache, V.; Xu, M.; Cadot, B.; Folker, E.S.; Richardson, B.E.; Gomes, E.R.; Baylies, M.K., MAP and kinesin-dependent nuclear positioning is required for skeletal muscle function. Nature, 2012. 484(7392): p. 120-124.
- Azevedo, M.; Baylies, M.K., Getting into Position: Nuclear Movement in Muscle Cells. Trends in Cell Biology, 2020. 30(4): p. 303-316.
- Romero, N.B., Centronuclear myopathies: A widening concept. Neuromuscular Disorders, 2010. 20(4): p. 223-228.
This entry is adapted from the peer-reviewed paper 10.3390/cells10020318