Despite great advances in the detailed profiling of tumor cells and the development of therapeutic agents, cancer metastasis is still a big hurdle in the treatment of cancer patients. This is possibly because tumor cells plastically evolve through interplay with the host environment, including stromal cells, vascular cells, and immune cells. The reciprocal evolution among the numerous components further increases the heterogeneity and complexity in both tumor cells and the host, leading to refractory cancer. It is important to better understand the entire metastatic cascade and the practical implementations targeting the oncoimmune drivers in the mechanisms.
- metastasis
- EMT
- cancer stem
- polyploidization
- immunity
- immunotherapy
- immune checkpoint
- immunosuppression
- immune exhaustion
- immune dysfunction
1. Introduction
Residual metastasis is a major cause of cancer-associated death. The epithelial–mesenchymal transition (EMT) is a pivotal biological program in cancer metastasis and confers high mobility, invasive, and metastatic properties, and anti-apoptotic dormancy on tumor cells [1][2], although several studies have reported the dispensability of EMT [3][4]. Great progress in the molecular profiling of cancer EMT and the related cancer-initiated cells or cancer stem cells (CSCs) has defined the detailed characteristics, and the advances in understanding the molecular basis of EMT have revealed the landscapes of cancer metastasis, leading to the development of numerous treatments targeting the phenotypes and signaling pathways [5].
To date, however, cancer metastasis is still a threat to cancer patients due to the high frequency and the high mortality in clinical settings. An important point is that the EMT is only a part of the metastatic cascade. The disseminated dormant tumor cells must reawaken to grow in the metastatic secondary site, possibly through the reverse process of EMT, the so-called mesenchymal-to-epithelial transition (MET), and/or polyploidy that giant cells rapidly generate progeny cells with genomic instability in response to treatment stress [6][7][8].
On the other hand, accumulating evidence suggests that epigenetic modification by the host environment, which is composed of numerous components, including stromal cells, vascular cells, and immune cells, impacts on every step of the metastatic cascade: tumor development, dissemination, invasion, intravasation, extravasation, colonization, and survival [9][10]. The reciprocal evolution among these factors could massively increase the oncoimmunological heterogeneity and complexity in the host, and raises the possibility of cancer metastasis. Advanced technologies have revealed the landscape of tumor-supportive immunity, and a variety of immunotherapeutics have been developed in clinical settings [11].
The most outstanding effort is targeting immune inhibitory checkpoint pathways, which are innate brakes on anti-tumor immune responses. Immune checkpoint inhibitors (ICIs) have been considered to be a promising immunotherapeutic to knockout the immune underpinnings and have shown great therapeutic efficacies, particularly long-lasting durable responses, even in patients with advanced and metastatic cancer [12]. However, the response rate remains relatively low in many cases since most patients eventually show acquired resistance to the treatment even if they respond in the beginning [12]. Adverse events, including autoimmunity [13] and hyperprogression [14][15], are frequently seen in the treated patients. Hyperprogression is a rapid deterioration of the tumor growth and metastasis in patients shortly after treatment with ICIs, although the incidence varies between 4% and 29% of all responses using various defining criteria [14][15]. Thus, more effective but less toxic treatments are still needed in clinical settings.
To disrupt the vicious spiral of the tumor–immunity aggravation leading to cancer metastasis, it is important to better understand both the entire metastatic cascade and the clinical implementation.
2. Treatments for Cancer Metastasis
Disruption of the cancer metastatic cascade is a promising strategy for treating cancer. Numerous agents targeting oncoimmune drivers, including small molecule inhibitors, antibodies, and genetically modified cells, have been pharmaceutically developed in clinical settings, as also reviewed elsewhere [16]. However, most of the clinical evaluations are still underway, and the therapeutic efficacy reported so far is limited to a subset of patients. Here, we summarize the recent advances in the development of agents as guidance for designing more effective treatment regimens in clinical settings (Figure 1).
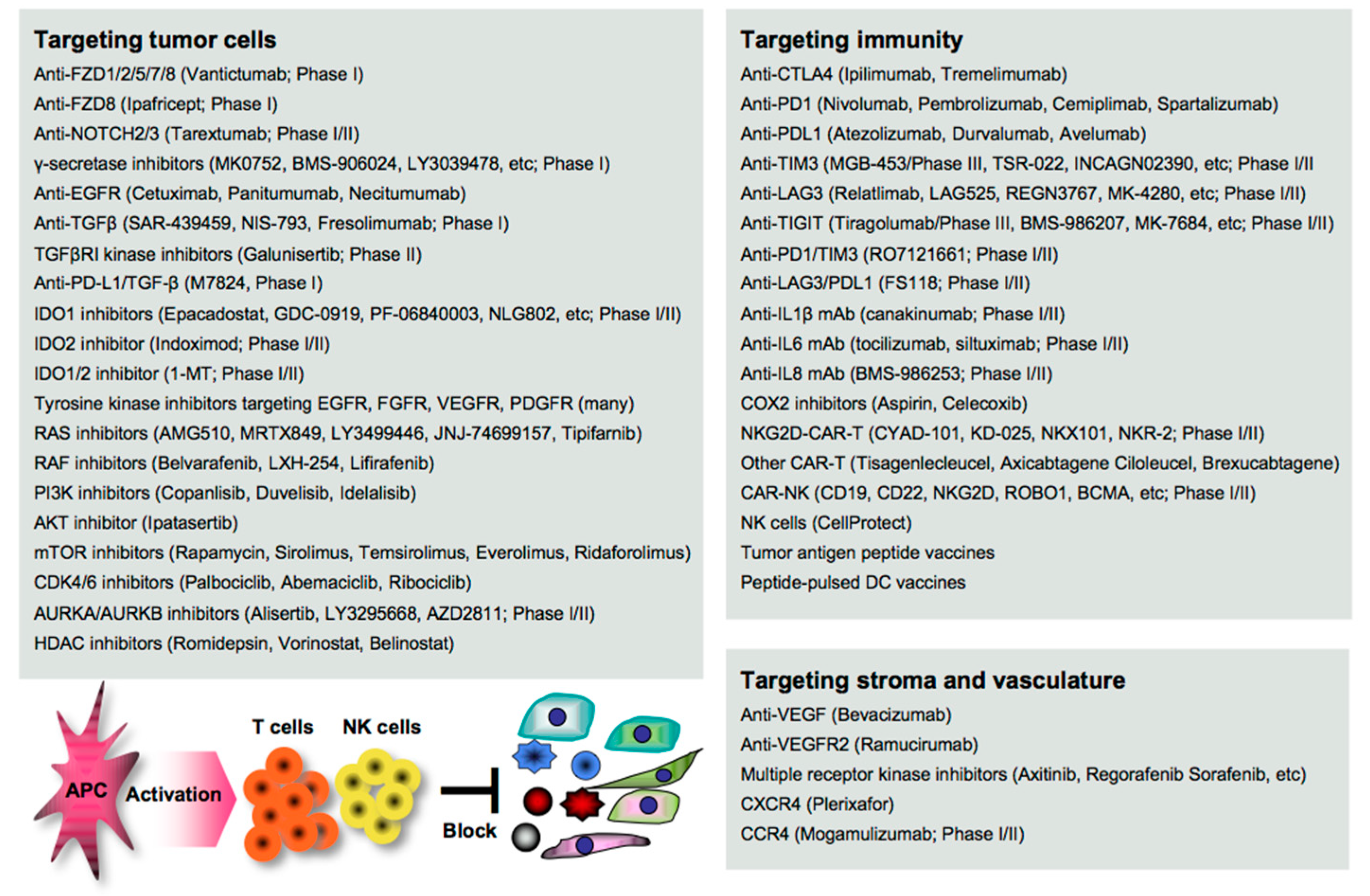
2.1. Targeting EMT/CSC Inducers
EMT stimuli simultaneously create a permissive environment for tumor escape in the host; thus, blocking the initiation of EMT appears to be a great rationale for the clearance of the complicated consequences. Vantictumab is an inhibitory mAb specific for the WNT receptors FZD1/2/5/7/8, which has been evaluated in a phase I study for solid tumors [17]. Ipafricept is a recombinant fusion protein with the extracellular domain of FZD8 and has been evaluated in combination with chemotherapy in a phase I study for advanced solid tumors [18]. Tarextumab (OMP-59R5) is an anti-NOTCH2/3 mAb and has been evaluated in phase I/II studies for solid tumors, including pancreatic cancer [19][20] γ-secretase is essential for NOTCH activation, and thus many inhibitors, including MK0752 [21], BMS-906024 [22], LY3039478 [23], RO4929097 [24], and LY900009 [25], have been evaluated in combination with/without chemotherapy in phase I studies for advanced or metastatic cancer.
Anti-EGFR mAbs (cetuximab, panitumumab, and necitumumab) have been clinically approved in combination with/without chemotherapy for various types of cancer [26][27]. Tyrosine kinases are essential for the activation of the signal transduction of receptors, and numerous inhibitors have been clinically developed and approved targeting EGFR (gefinitib, erlotinib, afatinib, simotinib, etc.), VEGFR (sunitinib, lenvatinib, vandetanib, etc.), FGFR (erdafitinib, infigratinib, pemigatinib, rogaratinib, etc.), PDGF (imatinib), and multiple receptors (axitinib, cabozatinib, ponatinib, regrorafenib, amcasertib, etc.) [28][29][30].
Blocking TGFβ and IDO is expected to widely organize the tumor environment, as both molecules have a broad range of biological activities on both tumor cells and host immunity as described above. Several anti-TGFβ mAbs (SAR-439459, NIS-793, and fresolimumab) have been evaluated in phase I studies for advanced solid tumors [31]. Galunisertib (LY2157299) is a small molecule inhibitor of TGFβ receptor I (TGFβRI) kinase for SMAD2 phosphorylation, and clinical outcomes have been reported in phase II studies for recurrent glioblastoma [32] and advanced hepatocellular carcinoma [33].
These TGFβ-targeting mAbs and inhibitors are often combined with anti-PD1/PDL1 therapy in clinical trials, as synergistic efficacy has been reported in mouse models [34]. M7824 is a bifunctional anti-PDL1/TGF-β trap fusion protein, which not only efficiently reverts the mesenchymalization of tumor cells, but also activates CTLs and NK cells [35], and many clinical trials have been conducted. Inhibitors targeting IDO1 (epacadostat, GDC-0919, PF-06840003, NLG802, SHR9146, and linrodostat), IDO2 (indoximod), or both (1-MT) have been evaluated in combination with chemotherapy and/or ICI therapy in phase I/II studies for solid tumors and peritoneal cancer [36]. In the phase 3 ECHO-301/KEYNOTE-252 study, however, the addition of epacadostat to pembrolizumab showed no significantly greater clinical benefit on overall survival and progression-free survival compared with pembrolizumab monotherapy in patients with unresectable or metastatic melanoma [37]. A reason may be that IDO and PDL1 expressions are generally low in melanoma tissues, as shown in open databases such as the Human Protein Atlas (http://www.proteinatlas.org/). The combination regimen may be effective in gastrointestinal cancer with higher expressions of both molecules.
RAS/RAF promotes the EMT through the MEK/ERK and the PI3K/AKT/mTOR signaling pathways [38], and thus numerous inhibitors have been clinically developed and approved targeting RAS (AMG510, MRTX849, LY3499446, JNJ-74699157, and tipifarnib), RAF (belvarafenib, LXH-254, and lifirafenib), PI3K (copanlisib, duvelisib, and idelalisib), AKT (ipatasertib), and mTOR (rapamycin, sirolimus, temsirolimus, everolimus, and ridaforolimus) for treating cancer patients [38][39].
Targeting the cell cycle-related CDK4/6/DUB3 pathway is also expected to contribute to the suppression of cancer metastasis, as blocking CDK4/6 and the related DUB3 is effective in mouse metastasis models [40][41]. CDK4/6 inhibitors (palbociclib, abemaciclib, and ribociclib) have been clinically approved and used in combination with/without hormone therapy for treating metastatic breast cancer [42]. Targeting polyploidy-related aurora kinases, including AURKA and AURKB, may also be useful for suppressing cancer metastasis as reported in mouse studies [43].
Many AURKA/AURKB inhibitors (alisertib, LY3295668, AZD2811, etc.) have been pharmaceutically developed [44]; however, most of the clinical trials failed. Six phase I/II studies have been now conducted in combination with chemotherapy and/or ICI therapy for various types of cancer (http://clinicaltrials.gov). The signaling network interacting with other signaling pathways is extremely complicated, and a number of feedback loops create refractory cancer. Therefore, combinations with certain agents could be better for treating cancer patients.
2.2. Targeting Stromagenesis and Angiogenesis
Blocking stroma-regulating drivers is also expected to impede tumor growth and dissemination. Despite the significant roles of MSCs and CAFs in cancer metastasis, directly targeting them is impractical because the precise characteristics are still obscure. Instead, blocking MSC/CAF-produced chemokines and the receptors may be an alternative approach. Many inhibitors have been pharmaceutically developed, and some targeting CXCR4 (plerixafor), CCR4 (mogamulizumab), and CCR5 (maraviroc) have been clinically approved, although only plerixafor is applicable for treating cancer, non-Hodgkin’s lymphoma, and multiple myeloma [45][46]. Mogamulizumab has now been evaluated in combination with anti-PD1 mAb (ClinicalTrials.gov Identifier: NCT03309878) or IL15 (ClinicalTrials.gov Identifier: NCT04185220) in phase I/II study for relapsed or refractory lymphoma and leukemia.
In contrast, targeting angiogenesis and vascularization has been progressed over time, and many small molecule inhibitors have been clinically approved targeting VEGF (bevacizumab), VEGFR2 (ramucirumab), and multiple receptors, including VEGFR, PDGFR, FGFR, KIT, RET, and FLT3 (axitinib, cabozantinib, nintedanib, pazopanib, regorafenib, sorafenib, and sunitinib) [26][27]. However, the anti-tumor efficacy of monotherapy is modest and limited, and combination regimens with other agents, including ICIs and chemotherapeutics, have now been evaluated in many clinical trials.
2.3. Targeting Immune Determinants
Immune mediators participate in the mechanisms underlying cancer metastasis. Blocking immune inhibitory pathways is important for inducing potent anti-tumor immunity, and targeting CTLA4/PD1 signaling has attracted great attention in cancer therapy [47]. There are many clinically approved mAbs targeting CTLA4 (ipilimumab and tremelimumab), PD1 (nivolumab, pembrolizumab, cemiplimab, and spartalizumab), and the ligand PDL1 (atezolizumab, durvalumab, and avelumab), and targeting the PD1-PDL1 axis has been widely recognized as a successful strategy for treating advanced and metastatic cancer, despite the clinical outcomes being extremely limited [12].
Biomarkers that predict potential responders to anti-PD1/PDL1 therapy have been strenuously investigated by analyzing patient-derived specimens using advanced technology, and several biomarkers, including PDL1 expression, microsatellite instability (MSI), and mutation burden (the number of non-synonymous single nucleotide variants) in tumor cells have been identified [48][49]. However, these are not necessarily correlated with clinical outcomes, and more precise and accurate biomarkers are still being explored in clinical settings. Combination regimens that potentially optimize the ICI efficacy have also been strenuously investigated, and numerous clinical trials with a variety of agents, such as small molecule inhibitors, ICIs, and vaccines, have been conducted around the world [50].
Targeting inflammatory mediators is also important for alleviating tumor aggravation and immune-related adverse events, including autoimmunity, which is frequently found in ICI therapy [13]. In general, inflammatory mediators have been pharmaceutically targeted primarily for treating other inflammatory diseases, such as rheumatoid arthritis and pulmonary disease, so far. However, several inhibitory mAbs have been clinically evaluated for treating cancer by targeting IL1β (canakinumab) [51], IL6 (tocilizumab, siltuximab) [52], and IL8 (BMS-986253) [53] in combination with/without other agents, such as chemotherapy, anti-HER2 mAb, or anti-PD1 mAb, in phase I/II trials.
COXs are representative inflammatory mediators that produce eicosanoids, such as PGE2 (mainly produced by COX2) and TXA2 (mainly produced by COX1) from arachidonic acid, and are highly and frequently expressed in both myeloid cells and tumor cells [54]. A number of preclinical studies demonstrated the therapeutic efficacy induced by a COX1/2 inhibitor aspirin on cancer metastasis by inhibiting platelet aggregation, endothelial activation, tumor cell adhesion to the endothelium, the recruitment of myeloid cells, and the EMT of tumor cells [55].
The clinical significance of aspirin use has also been demonstrated in colorectal cancer patients, particularly with PDL1low tumors [56][57]. However, most of the clinical studies are retrospective, and the therapeutic efficacies of COX inhibitors remain to be determined in clinical settings [58]. Interestingly, one study reported that treatment with a CDK4/6 inhibitor palbociclib suppressed COX2/PGE2 by the repression of c-JUN expression, resulting in the suppression of cancer metastasis in mouse breast cancer models [40]. Persistent and strong stimulation with inflammatory mediators induces exhaustion and dysfunction in anti-tumor effector cells, accompanied by the upregulation of multiple immune checkpoint molecules, including TIM3, LAG3, and TIGIT; therefore, blocking these negative signals is expected to reinvigorate the immune fighters against cancer [59]. Many inhibitory mAbs targeting TIM3 (TSR-022, MGB-453, INCAGN02390, Sym023, and BGB-A425), LAG3 (Relatlimab, LAG525, REGN3767, MK-4280, FS118, Syn-022, and TRS-003), and TIGIT (tiragolumab, BMS-986207, MK-7684, AB154, ASP8374, and COM902) have been clinically evaluated in combination with/without other agents, such as chemotherapeutics and ICIs in phase I/II trials and MGB-453 and tiragolumab in phase III trials (ClinicalTrials.gov: https://clinicaltrials.gov/).
This entry is adapted from the peer-reviewed paper 10.3390/cancers13030554
References
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84.
- Bakir, B.; Chiarella, A.M.; Pitarresi, J.R.; Rustgi, A.K. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020, 30, 764–776.
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-Mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476.
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-Mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530.
- Makena, M.R.; Ranjan, A.; Thirumala, V.; Reddy, A.P. Cancer stem cells: Road to therapeutic resistance and strategies to overcome resistance. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165339.
- Coward, J.; Harding, A. Size Does Matter: Why Polyploid Tumor Cells are Critical Drug Targets in the War on Cancer. Front. Oncol. 2014, 4, 123.
- Zhang, S.; Mercado-Uribe, I.; Xing, Z.; Sun, B.; Kuang, J.; Liu, J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene 2014, 33, 116–128.
- Amend, S.R.; Torga, G.; Lin, K.C.; Kostecka, L.G.; de Marzo, A.; Austin, R.H.; Pienta, K.J. Polyploid giant cancer cells: Unrecognized actuators of tumorigenesis, metastasis, and resistance. Prostate 2019, 79, 1489–1497.
- Liu, Y.; Cao, X. Immunosuppressive cells in tumor immune escape and metastasis. J. Mol. Med. 2016, 94, 509–522.
- Lorenzo-Sanz, L.; Munoz, P. Tumor-Infiltrating Immunosuppressive Cells in Cancer-Cell Plasticity, Tumor Progression and Therapy Response. Cancer Microenviron. 2019, 12, 119–132.
- Aguirre-Ghiso, J.A. How dormant cancer persists and reawakens. Science 2018, 361, 1314–1315.
- Popovic, A.; Jaffee, E.M.; Zaidi, N. Emerging strategies for combination checkpoint modulators in cancer immunotherapy. J. Clin. Invest. 2018, 128, 3209–3218.
- Varricchi, G.; Galdiero, M.R.; Marone, G.; Criscuolo, G.; Triassi, M.; Bonaduce, D.; Marone, G.; Tocchetti, C.G. Cardiotoxicity of immune checkpoint inhibitors. ESMO Open 2017, 2, e000247.
- Ferrara, R.; Mezquita, L.; Texier, M.; Lahmar, J.; Audigier-Valette, C.; Tessonnier, L.; Mazieres, J.; Zalcman, G.; Brosseau, S.; Le Moulec, S.; et al. Hyperprogressive Disease in Patients with Advanced Non-Small Cell Lung Cancer Treated With PD-1/PD-L1 Inhibitors or With Single-Agent Chemotherapy. JAMA Oncol. 2018, 4, 1543–1552.
- Frelaut, M.; Le Tourneau, C.; Borcoman, E. Hyperprogression under Immunotherapy. Int. J. Mol. Sci. 2019, 20, 2674.
- Stoletov, K.; Beatty, P.H.; Lewis, J.D. Novel therapeutic targets for cancer metastasis. Expert Rev. Anticancer Ther. 2020, 20, 97–109.
- Cho, E.S.; Kang, H.E.; Kim, N.H.; Yook, J.I. Therapeutic implications of cancer epithelial-mesenchymal transition (EMT). Arch. Pharm. Res. 2019, 42, 14–24.
- Jimeno, A.; Gordon, M.; Chugh, R.; Messersmith, W.; Mendelson, D.; Dupont, J.; Stagg, R.; Kapoun, A.M.; Xu, L.; Uttamsingh, S.; et al. A First-in-Human Phase I Study of the Anticancer Stem Cell Agent Ipafricept (OMP-54F28), a Decoy Receptor for Wnt Ligands, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 7490–7497.
- Smith, D.C.; Chugh, R.; Patnaik, A.; Papadopoulos, K.P.; Wang, M.; Kapoun, A.M.; Xu, L.; Dupont, J.; Stagg, R.J.; Tolcher, A. A phase 1 dose escalation and expansion study of Tarextumab (OMP-59R5) in patients with solid tumors. Investig. New Drugs 2019, 37, 722–730.
- Hu, Z.I.; Bendell, J.C.; Bullock, A.; LoConte, N.K.; Hatoum, H.; Ritch, P.; Hool, H.; Leach, J.W.; Sanchez, J.; Sohal, D.P.S.; et al. A randomized phase II trial of nab-paclitaxel and gemcitabine with tarextumab or placebo in patients with untreated metastatic pancreatic cancer. Cancer Med. 2019, 8, 5148–5157.
- Cook, N.; Basu, B.; Smith, D.M.; Gopinathan, A.; Evans, J.; Steward, W.P.; Palmer, D.; Propper, D.; Venugopal, B.; Hategan, M.; et al. A phase I trial of the gamma-secretase inhibitor MK-0752 in combination with gemcitabine in patients with pancreatic ductal adenocarcinoma. Br. J. Cancer 2018, 118, 793–801.
- Gavai, A.V.; Quesnelle, C.; Norris, D.; Han, W.C.; Gill, P.; Shan, W.; Balog, A.; Chen, K.; Tebben, A.; Rampulla, R.; et al. Discovery of Clinical Candidate BMS-906024: A Potent Pan-Notch Inhibitor for the Treatment of Leukemia and Solid Tumors. ACS Med. Chem. Lett. 2015, 6, 523–527.
- Massard, C.; Azaro, A.; Soria, J.C.; Lassen, U.; Le Tourneau, C.; Sarker, D.; Smith, C.; Ohnmacht, U.; Oakley, G.; Patel, B.K.R.; et al. First-in-Human study of LY3039478, an oral Notch signaling inhibitor in advanced or metastatic cancer. Ann. Oncol. 2018, 29, 1911–1917.
- Xu, R.; Shimizu, F.; Hovinga, K.; Beal, K.; Karimi, S.; Droms, L.; Peck, K.K.; Gutin, P.; Iorgulescu, J.B.; Kaley, T.; et al. Molecular and Clinical Effects of Notch Inhibition in Glioma Patients: A Phase 0/I Trial. Clin. Cancer Res. 2016, 22, 4786–4796.
- Pant, S.; Jones, S.F.; Kurkjian, C.D.; Infante, J.R.; Moore, K.N.; Burris, H.A.; McMeekin, D.S.; Benhadji, K.A.; Patel, B.K.R.; Frenzel, M.J.; et al. A first-in-Human phase I study of the oral Notch inhibitor, LY900009, in patients with advanced cancer. Eur. J. Cancer 2016, 56, 1–9.
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Zhang, S.; Gong, Z.; Li, X.; Cao, K.; Deng, H.; He, Y.; et al. The role of microenvironment in tumor angiogenesis. J. Exp. Clin. Cancer Res. 2020, 39, 204.
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell Mol. Life Sci. 2020, 77, 1745–1770.
- Jiao, Q.; Bi, L.; Ren, Y.; Song, S.; Wang, Q.; Wang, Y.S. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol. Cancer 2018, 17, 36.
- Huang, L.; Jiang, S.; Shi, Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J. Hematol. Oncol. 2020, 13, 143.
- Facchinetti, F.; Hollebecque, A.; Bahleda, R.; Loriot, Y.; Olaussen, K.A.; Massard, C.; Friboulet, L. Facts and New Hopes on Selective FGFR Inhibitors in Solid Tumors. Clin. Cancer Res. 2020, 26, 764–774.
- Yang, X.G.; Zhu, L.C.; Wang, Y.J.; Li, Y.Y.; Wang, D. Current Advance of Therapeutic Agents in Clinical Trials Potentially Targeting Tumor Plasticity. Front. Oncol. 2019, 9, 887.
- Brandes, A.A.; Carpentier, A.F.; Kesari, S.; Sepulveda-Sanchez, J.M.; Wheeler, H.R.; Chinot, O.; Cher, L.; Steinbach, J.P.; Capper, D.; Specenier, P.; et al. A Phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro Oncol. 2016, 18, 1146–1156.
- Kelley, R.K.; Gane, E.; Assenat, E.; Siebler, J.; Galle, P.R.; Merle, P.; Hourmand, I.O.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; et al. A Phase 2 Study of Galunisertib (TGF-β1 Receptor Type I Inhibitor) and Sorafenib in Patients With Advanced Hepatocellular Carcinoma. Clin. Transl. Gastroenterol. 2019, 10, e00056.
- Principe, D.R.; Park, A.; Dorman, M.J.; Kumar, S.; Viswakarma, N.; Rubin, J.; Torres, C.; McKinney, R.; Munshi, H.G.; Grippo, P.J.; et al. TGFβ Blockade Augments PD-1 Inhibition to Promote T-Cell-Mediated Regression of Pancreatic Cancer. Mol. Cancer Ther. 2019, 18, 613–620.
- Jochems, C.; Tritsch, S.R.; Pellom, S.T.; Su, Z.; Soon-Shiong, P.; Wong, H.C.; Gulley, J.L.; Schlom, J. Analyses of functions of an anti-PD-L1/TGFβR2 bispecific fusion protein (M7824). Oncotarget 2017, 8, 75217–75231.
- Zhu, M.M.T.; Dancsok, A.R.; Nielsen, T.O. Indoleamine Dioxygenase Inhibitors: Clinical Rationale and Current Development. Curr. Oncol. Rep. 2019, 21, 2.
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097.
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-Targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552.
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26.
- Qin, G.; Xu, F.; Qin, T.; Zheng, Q.; Shi, D.; Xia, W.; Tian, Y.; Tang, Y.; Wang, J.; Xiao, X.; et al. Palbociclib inhibits epithelial-mesenchymal transition and metastasis in breast cancer via c-Jun/COX-2 signaling pathway. Oncotarget 2015, 6, 41794–41808.
- Wu, Y.; Wang, Y.; Lin, Y.; Liu, Y.; Wang, Y.; Jia, J.; Singh, P.; Chi, Y.I.; Wang, C.; Dong, C.; et al. Dub3 inhibition suppresses breast cancer invasion and metastasis by promoting Snail1 degradation. Nat. Commun. 2017, 8, 14228.
- Goel, S.; DeCristo, M.J.; McAllister, S.S.; Zhao, J.J. CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest. Trends Cell Biol. 2018, 28, 911–925.
- Kozyreva, V.K.; Kiseleva, A.A.; Ice, R.J.; Jones, B.C.; Loskutov, Y.V.; Matalkah, F.; Smolkin, M.B.; Marinak, K.; Livengood, R.H.; Salkeni, M.A.; et al. Combination of Eribulin and Aurora a Inhibitor MLN8237 Prevents Metastatic Colonization and Induces Cytotoxic Autophagy in Breast Cancer. Mol. Cancer Ther. 2016, 15, 1809–1822.
- Bavetsias, V.; Linardopoulos, S. Aurora Kinase Inhibitors: Current Status and Outlook. Front. Oncol. 2015, 5, 278.
- Vela, M.; Aris, M.; Llorente, M.; Garcia-Sanz, J.A.; Kremer, L. Chemokine receptor-specific antibodies in cancer immunotherapy: Achievements and challenges. Front. Immunol. 2015, 6, 12.
- Miao, M.; De Clercq, E.; Li, G. Clinical significance of chemokine receptor antagonists. Expert Opin. Drug Metab. Toxicol. 2020, 16, 11–30.
- Arasanz, H.; Gato-Canas, M.; Zuazo, M.; Ibanez-Vea, M.; Breckpot, K.; Kochan, G.; Escors, D. PD1 signal transduction pathways in T cells. Oncotarget 2017, 8, 51936–51945.
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150.
- Park, Y.; Koh, J.; Na, H.Y.; Kwak, Y.; Lee, K.W.; Ahn, S.H.; Park, D.J.; Kim, H.H.; Lee, H.S. PD-L1 Testing in Gastric Cancer by the Combined Positive Score of the 22C3 PharmDx and SP263 Assay with Clinically Relevant Cut-offs. Cancer Res. Treat. 2020, 52, 661–670.
- Xin Yu, J.; Hubbard-Lucey, V.M.; Tang, J. Immuno-Oncology drug development goes global. Nat. Rev. Drug Discov. 2019, 18, 899–900.
- Schenk, K.M.; Reuss, J.E.; Choquette, K.; Spira, A.I. A review of canakinumab and its therapeutic potential for non-small cell lung cancer. Anticancer Drugs 2019, 30, 879–885.
- Rossi, J.F.; Lu, Z.Y.; Jourdan, M.; Klein, B. Interleukin-6 as a therapeutic target. Clin. Cancer Res. 2015, 21, 1248–1257.
- Bilusic, M.; Heery, C.R.; Collins, J.M.; Donahue, R.N.; Palena, C.; Madan, R.A.; Karzai, F.; Marte, J.L.; Strauss, J.; Gatti-Mays, M.E.; et al. Phase I trial of HuMax-IL8 (BMS-986253), an anti-IL-8 monoclonal antibody, in patients with metastatic or unresectable solid tumors. J. Immunother. Cancer 2019, 7, 240.
- Stasinopoulos, I.; Shah, T.; Penet, M.F.; Krishnamachary, B.; Bhujwalla, Z.M. COX-2 in cancer: Gordian knot or Achilles heel? Front. Pharmacol. 2013, 4, 34.
- Lucotti, S.; Cerutti, C.; Soyer, M.; Gil-Bernabe, A.M.; Gomes, A.L.; Allen, P.D.; Smart, S.; Markelc, B.; Watson, K.; Armstrong, P.C.; et al. Aspirin blocks formation of metastatic intravascular niches by inhibiting platelet-derived COX-1/thromboxane A2. J. Clin. Invest. 2019, 129, 1845–1862.
- Tougeron, D.; Sha, D.; Manthravadi, S.; Sinicrope, F.A. Aspirin and colorectal cancer: Back to the future. Clin. Cancer Res. 2014, 20, 1087–1094.
- Hamada, T.; Cao, Y.; Qian, Z.R.; Masugi, Y.; Nowak, J.A.; Yang, J.; Song, M.; Mima, K.; Kosumi, K.; Liu, L.; et al. Aspirin Use and Colorectal Cancer Survival According to Tumor CD274 (Programmed Cell Death 1 Ligand 1) Expression Status. J. Clin. Oncol. 2017, 35, 1836–1844.
- Jin, M.Z.; Jin, W.L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target Ther. 2020, 5, 166.
- Pauken, K.E.; Wherry, E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015, 36, 265–276.