Caffeic acid phenethyl ester (CAPE), is one of the most extensively investigated active components of propolis and it is considered responsible for most of its beneficial effects. Among the others, CAPE exerts protection towards many neurological disorders. This review summarizes the protective effects of CAPE towards oxidative stress, inflammation, apoptosis, neurotoxic substances, neurodegenerative diseases, brain tumors and neuronal injuries as well. A paragrah on derivatives of CAPE is also included.
- Caffeic Acid Phenethyl Ester
- neuroprotection
- neurodegenerative disease
- propolis
- CNS injury
- CNS ischemia
- CNS cancer
- neuroinflammation
- CAPE derivatives
- neurotoxic substance(s)
- Introduction
Propolis, a honeybee product, is a resinous product obtained from different plant parts mixed with beeswax and bee salivary enzymes, and represents a market of multimillion dollar [1,2]. It has been used in traditional medicine for many years due to its anti-microbial, anti-inflammatory, anti-oxidant, immunomodulator, anti-mutagenic, and carcinostatic effects [3]. Propolis consists of a large number of different compounds according to the ingredients the bees collect from the plants. Its main components are phenols and related compounds. Investigations have been conducted to purify and determine which substances are responsible for these effects. The results of these investigations suggested that caffeic acid phenethyl ester (CAPE, phenethyl 3-(3-4 dihydroxyphenyl) acrylate) is the main component that mediates most of the beneficial effects ascribed to the propolis [4]. As a consequence, CAPE has become one of the most studied natural product research topics in recent years. Grunberger and co-workers extracted and described CAPE in 1988[5], and later Sigma-Aldrich produced commercial preparations for the market [5]. CAPE is a polyphenol with hydroxyl groups within the catechol ring which provide strong antioxidant properties to the molecule and can affect many biological activities. It has a lipophilic property because of its long carbon groups in an aromatic and aliphatic structure which leads to adequate blood concentration after intraperitoneal administration [6]. Celli et al. [8] investigated the stability of CAPE in rats and human plasma. It was found that CAPE undergoes hydrolysis to caffeic acid after 6 h in rat plasma in vitro and also in vivo giving caffeic acid as the major metabolite. This type of hydrolysis does not occur in human plasma because it does not contain the carboxylesterase, which is responsible for the CAPE hydrolysis [7]. The broad spectrum of pharmacological activities shown by CAPE ranges from anti-oxidant/anti-inflammatory to anti-viral/anti-fungal properties, including anti-proliferative effects in various cancer models [8]. A recent in vivo study indicated that CAPE has the ability to cross the blood–brain barrier in rats [7]. This review focuses on the protective effects of CAPE in many diseases that can affect the central nervous system (CNS). CAPE was found to have a protective effect on different neurodegenerative disorders, either occurring with age, like Alzheimer’s disease (AD) and Parkinson’s disease (PD), or other neurodegenerative disorders, as amyotrophic lateral sclerosis (ALS) and seizures. CAPE proved to protect neurons from the main underlying causes of several human neurologic diseases, namely oxidative stress, apoptosis dysregulation and brain inflammation. In addition, CAPE was found able to protect the nervous system from the consequences of some diseases not directly affecting it, like diabetes, septic shock and hepatic encephalopathy (HE). A paragraph about the neuroprotective effects of CAPE against adverse reactions induced by different neurotoxic substances like ethanol, methotrexate, acrolein and others is also provided. The role of CAPE in the pathophysiology of CNS tumors and in neuronal injuries is carefully reviewed through the description of the modulatory activity of CAPE against biochemical and histopathological cascade mechanisms following ischaemic conditions in the brain and spinal cord. Finally, a brief overview of CAPE synthetic derivatives follows. Five tables summarize the main findings on CAPE effects in different areas of neuroprotection. In the column named “Parameters measured” the effect of CAPE was reported between parenthesis where applicable. In some case, the effect of CAPE is not directly deducible, but it depends on the experimental model or conditions. This review provides a useful insight into the role of CAPE in the management of numerous pathological conditions affecting the CNS. The description of the molecular mechanisms and the focus on CAPE derivatives can help in discovering new promising targets for neuroprotection and in finely tailoring structural modifications of CAPE derivatives.
- Methodology
Online database PubMed was used for screening the studies for this review with no chronological limits applied. Keywords used to search for articles included caffeic acid phenylethyl ester and neuroprotection, neurodegenerative disorders, CNS injury, CNS ischaemia, CNS cancer, neuroinflammation, brain injury, spinal cord injury and neuronal injury. The search retrieved more than 90 results, including three reviews with a section or hints about neuroprotective effect of CAPE or its protective effects against ischemia-reperfusion injury. None of these discussed the diverse signaling pathways involved. After having removed duplicates and narrowed the results according to the relevance to the core topic of the review, 77 papers were selected. The total of 90 references was reached with the inclusion of papers useful to better introduce and clarify the subjects of the six paragraphs.
- CAPE effects on different neurologic disorders
With age a lot of neurologic disorders are common to occur such as Alzheimer’s disease (AD), Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS). Common causes for the development of these diseases are oxidative stress, inflammation and apoptosis impairment. A main goal of anti-ageing strategies is the elimination of harmful agents, in other words free radicals, from the environment to protect organs from aging and other oxidative stress-related pathologies. One study evaluated the effects of long term administration of CAPE on histological and biochemical alterations induced by ageing in brain and cerebellum of old rats. CAPE increased superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GSH-Px) activities and glutathione (GSH) levels in both cerebral and cerebellar tissues to levels similar to those in young rats. Additionally, CAPE reduced malondialdehyde (MDA, an important marker of lipid peroxidation) level and ageing-induced ultrastructural alterations, proving to be more effective than melatonin, which is used worldwide in order to prevent ageing related pathologies [9]. Many studies addressing the protective effects of CAPE on different neurologic disorders are described below and summarized in Table 1. Other than the above mentioned pathological conditions, the role of CAPE in psychosis, seizures and the side-effects of other diseases are described.
3.1. Apoptosis
Apoptosis dysregulation has been suggested to be the underlying cause of several human neurologic diseases. Transient focal or global cerebral ischaemia in rodents leads to neuronal apoptosis. Reactive oxygen species (ROS) production is thought to represent a relevant mechanism in the series of biochemical events ultimately leading to apoptosis. CAPE blocks neuronal death through inhibition of inflammation and of mitochondrial cytochrome c release, and through reduction of free radical generation [10,11]. Primary cultures of cerebellar granule neurons (CGNs) are considered a suitable in vitro model to study the mechanisms of neuronal apoptosis, which could be induced by low K+ concentrations. CAPE exerts its anti-apoptotic effect on CGNs by blocking ROS formation and by inhibiting the activity of both caspase-3 and caspase-9. Caspase-3 is the executioner caspase that is downstream of the intrinsic (mitochondrial) caspase-9 and extrinsic receptor activated (Fas) caspase-8 pathways. Interestingly, CAPE was found to completely block the activation of the nuclear factor-κB (NF-κB) without interfering with the marked decrease in intracellular Ca2+ concentration induced by low K+ [10,12].
Another study investigated CAPE neuroprotective effect against apoptotic cell death in the developing rat brain after pentylenetetrazole (PTZ)-induced status epilepticus (SE). Prolonged seizures are associated with inadequate blood flow, increased excitatory amino acid release, and decreased glucose use and oxygen consumption. All these features are associated with impaired mitochondrial function and irreversible neuronal damage. CAPE showed diminished apoptosis and preservation of the number of neurons, as well as a reduced number of caspase-3 positive cells in the hippocampus and in the prefrontal cortex [13]. Another study [14] investigated the effect of CAPE on CGNs exposed to excitotoxicity by overstimulation of glutamate receptors. Neuronal death caused by excitotoxicity plays an important role in a number of neurodegenerative disorders, including Alzheimer’s disease, Parkinson’s disease and multiple sclerosis. CAPE inhibited necrosis mediated by p38 phosphorylation and apoptosis mediated by cytochrome c release and caspase-3 activation, while appeared to have no effect on glutamate-induced neuronal death via the NF-κB pathway. Interestingly, CAPE proved effective in modulating neurotogenesis, i. e. the formation of new neurites, in PC12 cells challenged with the dopaminergic neurotoxin 1-methyl-4-phenylpyridinium (MPP+). CAPE induced the formation, elongation and ramification of neurites and inhibited the shortage of neurites induced by the neurotoxin. These effects were associated with increased expression of neuronal typical proteins responsible for axonal growth (GAP-43) and synaptogenesis (synaptophysin and synapsin I) [15].
3.2. Neuro-inflammation
Brain inflammatory events are observed in several neuropathologies or in response to infectious diseases. CAPE’s ability to control neuro-inflammation was compared with other drugs such as acetyl-salicylate (anti-inflammatory), dexamethasone (glucocorticoid), pyrrolidine dithiocarbamate (anti-oxidant) and SN 50 peptide (selective permeant NF-κB inhibitor) in organotypic hippocampal slice cultures. Interestingly, CAPE presented a longer lasting control (over 48 h after a single administration) of neuro-inflammation compared to the other compounds. This effect can be ascribed to interference CAPE exerts on several neuroinflammatory effectors, such as NF-κB, tumor necrosis factor (TNF)-α and nitric oxide (NO). At the dose of maximal effect (100 µM), an increase in cAMP-responsive element binding protein (CREB) activity, a molecule with anti-inflammatory activity, was also observed [16]. Another study examining the anti-inflammatory effect of CAPE on activated astroglial cells, found that the modulation of NF-κB activity in astroglial cells can be a potential target for treatment of inflammatory and degenerative CNS diseases. CAPE pre-treatment abrogated TNF-α induced expression of chemokine (C-C motif) ligand 2 (CCL-2) and intercellular adhesion molecule-1 (ICAM-1) via inhibition of NF-κB activation in a cell-specific manner. CAPE inhibited downstream pathways of inhibitor κB (IκB) degradation in monocytic cells, while suppressing the activation of upstream IkB kinase in astroglial cells [17].
X-linked adrenoleukodystrophy (X-ALD) is a neuro-inflammatory disease associated with demyelination of the cerebral white matter, characterized by accumulation of very long chain fatty acids (VLCFA), caused by peroxisomal disorder due to mutations in the ABCD1 gene. CAPE treatment corrected both the metabolic VLCFA accumulation and the secondary inflammation. The effect of CAPE was mediated by upregulation of Abcd2 expression and of peroxisomal β-oxidation, leading to decreased VLCFA levels in ABCD1-deficient U87 cells. CAPE administration reduced the expression of inducible nitric oxide synthase (iNOS), inflammatory cytokines, and the activation of NF-κB, in primary astrocytes derived by ABCD1/ABCD2-silenced mice [18]. Inflammation is a major component in the pathogenesis of another disease; experimental autoimmune encephalomyelitis (EAE), which is considered the animal model of multiple sclerosis (MS). In both EAE and MS, ROS can damage the myelin sheath and the blood–brain barrier (BBB). CAPE maintains cell membrane integrity and function, thus preventing protein leakage and accumulation by inhibiting peroxidation of membrane lipids. CAPE may exert its anti-inflammatory effect and ameliorate clinical symptoms by inhibiting ROS production at the transcriptional level, through the suppression of NF-κB activation, and by directly inhibiting the iNOS catalytic activity [19].
ALS is a disease that causes the death of motor neurons controlling voluntary muscles. In one study in 2009 [20] CAPE was selected from a library of 2000 small anti-oxidant molecules for the mechanism of action and the ability to be in the CNS in a concentration sufficient to give a therapeutic effect. In two experimental models of ALS, represented by mouse motor neuron cells (NSC34) expressing mutant superoxide dismutase 1 (SOD1) and motor neurons isolated from cases of familial SOD1-associated ALS, CAPE inhibited NF-κB-induced inflammation. CAPE also activated the nuclear factor erythroid 2-related factor 2 (Nrf2)–ARE pathway, which is usually down-regulated in ALS. Genes up-regulated by Nrf2 include the GSH-Px, glutathione reductase, the heme oxygenase 1 (HO-1); enzymes involved in GSH synthesis; and NADPH-regenerating enzymes. CAPE effects were tested also in an in vivo model of mice expressing a mutant superoxide dismutase (SOD1G93A) linked to human ALS. CAPE increased the post-onset survival and lifespan of the mice, showing a significantly greater number of surviving motor neurons and a decreased number of activated microglia and astrocytes in the lumbar region of the spinal cord. In addition, lower levels of phosphorylated p38, a mitogen-activated protein kinase that is involved in both inflammation and neuronal death, were observed in the spinal cords of SOD1G93A mice [21]. Microglial activation has been widely demonstrated to mediate inflammatory processes that are crucial in several neurodegenerative disorders. CAPE proved to inhibit cyclooxygenase-2 (COX-2) and iNOS expression and the consequent NO production both in an in vitro and in vivo models of microglia activation. Anti-neuroinflammatory responses in microglial cells were mediated by 5'-adenosine monophosphate-activated protein kinase (AMPK)α, erythropoietin (EPO), and HO-1 [22].
3.3. Parkinson’s disease
PD is a neurodegenerative disorder characterized by the progressive loss of dopaminergic neurons of the substantia nigra pars compacta. 6-Hydroxydopamine (6-OHDA) is a neurotoxic synthetic organic compound used to selectively destroy dopaminergic neurons in the brain to induce PD in laboratory animals. Neurotoxicity and neuronal cell death induced by 6-OHDA is due to ROS production, cytochrome c release and subsequent caspase-3 activation. CAPE increased the viability of cerebellar granule neurons dose dependently, and markedly attenuated 6-OHDA-induced toxicity. CAPE blocked Ca2+-induced cytochrome c release and caspase-3 activation but no interaction of CAPE with caspase-3 cleavage was found [23]. These results are confirmed in the same cellular model and in rostral mesencephalic neurons (RMNs), where CAPE pretreatment increased neuronal viability from 39% to 91% in CGNs and from 33% to 60% in RMNs. CAPE is able to block 6-OHDA-induced dopaminergic neuronal death through blockage of O2−· and peroxynitrite generation, suggesting its potential use as aneuroprotective drug for PD [24]. Coming to the in vivo experimental models, CAPE inhibited mitochondrial permeability transition, a process that triggers cytochrome c release and caspase-3 activation, leading to neuronal death in rats treated with 6-OHDA to induce PD. The mechanism of protection involves scavenging of free radicals and metal chelation without mitochondrial dysfunction [25].
CAPE can protect midbrain dopaminergic neurons in vitro and in vivo also by induction of HO-1 and brain-derived neurotrophic factor (BDNF) expression [26]. Rotenone is commonly used to induce experimental PD because of its ability to block the complex I inhibition and to activate microglia, resulting in neuropathologic and phenotypic features of PD. In a rotenone Parkinsonian mice model, CAPE treatment raised the level of striatal dopamine and lessened the inflammatory burden, by suppressing microglia cells and down regulating the gene expression of COX-2, iNOS and NF-κB. These effects led to improvement in locomotor activity. CAPE exposure protected against rotenone-induced histopathological abnormalities and led to an increased number of surviving neurons as well [27]. In a model for PD induced by Chlorpyrifos, CAPE showed positive effects on paraoxonase (PON1) activity, levels of lipid profile, total sialic acid (TSA), total anti-oxidant capacity (TAC) and total oxidant capacity (TOC) in plasma and brain tissue, preventing neurodegeneration [28]. In a murine model of PD induced by MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), CAPE attenuated dopaminergic neurodegeneration and dopamine loss. This effect was associated with a marked reduction of iNOS and caspase-1 expression in vivo. In vitro, CAPE proved able to mitigate neurotoxicity by inhibiting the mitochondrial release of cytochrome c and apoptosis inducing factor (AIF) induced by MPP+ [29].
3.4. Alzheimer’s disease
Alzheimer’s disease (AD) is an age-associated neurodegenerative disease that can be induced in rodents by intracerebroventricular (ICV) administration of a widely used diabetogenic drug, streptozotocin (STZ). Brain PI3-kinase activity and nitric oxide production mediated by the endothelial NOS (eNOS) are involved in the memory revival function of CAPE in STZ-ICV administered rats. Basal NF-κB activity in rat brain was found essential for memory functions as well. The anti-apoptotic and pro-survival functions of PI3-kinase activation regulate eNOS and NFκB activity, conferring neuroprotection and improving memory [30]. A previous study by the same group showed that CAPE down-regulation of oxidative stress and inflammation was accompanied by amelioration of STZ-ICV-induced dementia . A great increase in brain GSH levels and a diminution of thiobarbituric acid reactive substances (TBARS), as well as of TNF-α content, were observed in brains of rats treated with CAPE [31]. In another study amyloid-beta oligomers were administered to mice to induce dementia and to study the AD onset and the cognitive function impairment. CAPE treatment reversed cognitive deficits and improved learning and memory abilities. This action was accompanied by an induction of Nrf2 and heme oxygenase-1 via the modulation of glycogen synthase kinase 3β in the murine hippocampus [32].
3.5. Seizures and psychosis
CAPE is useful as an adjunctive treatment of seizure disorders. Seizures induced by pentylenetetrazole (PTZ) are due to the activation of glutamate receptors and the inhibition of GABA, an inhibitory neurotransmitter. Glutamate receptors’ activation enhances ROS level, which in turn enhance glutaminergic activity, but the administration of CAPE increased the latency and decreased the duration of seizures in PTZ-treated mice. CAPE protected the brain tissue from oxidative damage because of its ability to scavenge ROS, to decrease MDA concentration, to increase anti-oxidant SOD level and to significantly attenuate NO generation [11].
Psychosis has many different causes and can be experimentally induced by dizocilipine maleate (MK-801) which causes neurotoxicity inducing intracellular generation of free radical species through the N-methyl-D-aspartate (NMDA) receptor blockage. NMDA antagonists mediate cell death triggered by ROS, which are involved in membrane pathology in CNS and play a role in neuropsychiatric disorders, including schizophrenia. In one study focusing on rat prefrontal cortex (PFC), which is the region mainly affected in schizophrenia, CAPE modulated the brain oxidant/anti-oxidant status, stabilizing the cellular membranous structures. CAPE therapeutic effect was exerted by decreasing the levels of malondialdehyde, protein carbonyl (PC) and nitric oxide (NO). The activity of the enzymes glutathione peroxidase (GSH-Px), xanthine oxidase (XO) and adenosine deaminase (ADA) in prefrontal tissue where decreased as well, when compared to MK-801 groups, whereas catalase activity was not changed. In addition, CAPE treatment decreased the number of apoptotic cells in the PFC exposed to MK-801 [33]. Figure 1 summarizes the molecules involved in the effects exerted by CAPE in different neurological conditions described in the previous paragraphs.
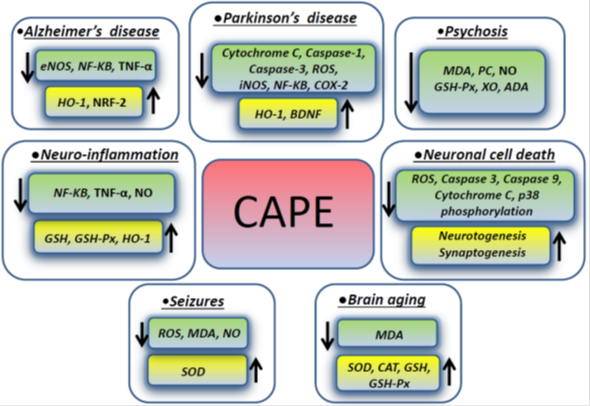
Figure 1. CAPE effects on intracellular molecules and pathways in different neurologic disorders
3.6. Other diseases
Some pathological conditions, different from neurodegenerative disorders, like diabetes, septic shock and hepatic encephalopathy (HE), can affect the health of the nervous system. Diabetes induces oxidative stress and inflammation in brain. In a murine model, CAPE significantly counteracted the effects of diabetes by decreasing the levels of nitric oxide and malondialdehyde, and the activities of catalase, glutathione peroxidase, and xanthine oxidase. CAPE treatment significantly suppressed also the expression of inflammatory cytokines such as TNFα and interferon (IFN)-γ and the iNOS activity, which were remarkably enhanced in brain by diabetes [34].
Sepsis patients suffer from severe oxidative stress with the overproduction of reactive oxygen species and reactive nitrogen species , resulting in direct cellular injury. NF-κB plays a central role in the induction of crosstalk between cytokines and inflammatory mediators, which leads to the pathophysiology of septic shock. The effect of CAPE against lipopolysaccharide (LPS) induced endotoxemia, neuronal damage and the associated systemic inflammatory response was investigated in male Wistar rats. CAPE prevented neuronal damage and preserved astrocyte morphology, with no sign of inflammatory cellular infiltration, and of edema or cytoplasmic swelling. CAPE decreased the levels of inflammatory cytokines (interleukin (IL)-1α, -1β, -6) and of the TNF-α in the plasma, increased the anti-inflammatory cytokines levels (IL-4, IL-10) and counteracted the imbalance leading to the inhibition of adhesion molecule expression (sICAM-1). This effect of CAPE on the inflammatory cellular infiltration into the brain could be directly attributed to its inhibitory effect on NF-κB activation [35].
Hepatic encephalopathy (HE) is a major neurological complication secondary to severe liver failure causing serious neurological problems. Thioacetamide-induced HE in rats was almost fully reversed by a combination of CAPE with the laxative lactulose. The survival rates were 37.5% in HE group, 70% in HE + lactulose group, 80% in HE + CAPE group, and 100% in HE + CAPE + lactulose group. The lack of death in the animals treated with both CAPE and lactulose can be ascribed to the direct neuroprotective effect of CAPE together with the prevention of ammonia production in the body. Increased ammonia, high transaminase levels in blood, increased lipid peroxidation and decreased antioxidant enzyme activities in most brain regions, along with impaired sensorymotor behavioural tests, were reversed to almost control values in CAPE + lactulose treated group [36].
Glaucoma is characterized by the death of retinal ganglion cells (RGCs) and visual field defects leading to irreversible blindness. CAPE prevented optic nerve crush-induced RGC apoptosis and neuroinflammation. These effects are mediated by the decreased expression of the inflammatory cytochines IL-8 and IL‑6, inducible nitric oxide synthase, cycloooxygenase‑2, tumor necrosis factor‑α and chemokine C‑C ligand‑2. Hypertrophy of astrocytes and Müller cells (gliosis) was modulated by CAPE through the inhibition of NF‑κB signaling [37].
Table 1: CAPE protective effects in different neurological disorders. Where applicable the effect of CAPE on the parameters measured is reported between parenthesis (+, - or =).
|
Stimulus |
Parameters measured |
CAPE dose(s) |
Experimental model |
Ref. |
|
Aging: 18 month old rats |
-Histopathological assessment -MDA (-) -SOD (+) -CAT (+) -GSH-Px (+) -GSH (+) |
15 mg/kg/day, i.p, for 95 days |
Male Sprague Dawley rats |
[9] |
|
Apoptosis: serum-free medium with low K+ (5mM KCl) |
-Apoptosis (-) -ROS (-) -Ca2+ (=) -NF-κB (-) -Caspase-3 and -9 (-) |
10µg/ml |
Primary CGNs from 8-day-old Wistar rats |
[10] |
|
SE: PTZ 40-mg/kg followed by 10 mg/kg every 10 min until SE occurrence, i.p. |
-Histopathological assessment -Caspase-3 (-) |
30 mg/kg /day, i.p, for 5 days starting 40 min after the SE tonic phase |
Dams reared Wistar male rats |
[13] |
|
Excitotoxicity: -Cells: glutamate 30 µM/24 h -Isolated mitochondria: glutamate and maleate (both 5 mM) |
-Cell viability (+) -Caspase-3 (-) -Cytochrome c (-) -Glutamate-evoked currents |
0 µM -200 µM, pre- and co-treatment |
-CGNs from 8-day-old Sprague Dawley rats -Mitochondria from CGNs and livers |
[14] |
|
Cytotoxicity: MPP+ 100, 500 or 1000 µM |
-Cell differentiation (+) -Cell viability (+) -Protein content (+) -Synaptophysin (+) -GAP-43 (+) -Synapsin I (+) |
1, 5 or 10 µM |
PC-12 cells |
[15] |
|
Neuroinflammation: IFN-γ and LPS
|
-NF-κB (-) -TNF-α (-) -NOS-2 (iNOS) (-) -CREB (+) |
4 to 100 µM, 30 min before and during LPS exposure |
Organotypic hippocampal cultures from the hippocampi of 5–7- day-old Wistar rats |
[16] |
|
Neuroinflammation: TNF-α 10 ng/mL/6 h |
-CCL-2 (-) -CXCL-8 (-) -ICAM-1 (-) -Monocyte Adhesion (-) -DNA-binding activity of NF-κB and AP-1 (-) -IκBα -IKK -TRAF2 -TAK1 -MKK4 -JNKs -c-Jun |
30 µM, pretreatment |
-CRT-MG human astroglial cells -U937 human monocytic cells |
[17] |
|
XALD: human skin fibroblasts derived from XALD (GM04932, GM04934), and AMN (GM07531) patients |
-TNF-α (-) -ROS (-) -NO (-) -Fatty acids (-) |
1-5µM |
-Fibroblasts -Mouse primary mixed glia and astrocytes |
[18] |
|
EAE: 50 µg of guinea pig MBP and 7 mg/ml heat-killed Mycobacterium tuberculosis, intradermally. |
-Neurological assessment -MDA (-) -NO (-) -XO -GSH-Px -ADA -SOD |
25 µmol/kg/day, i.p. for 14 days after immunization |
Female Wistar rats |
[19] |
|
ALS: transfection with pIRESneo and/or SOD1 mutants |
-In silico analysis -DCF -Cell viability (+) -Nrf2 (+) -5- LO -NF-κB (-) |
10µM, co-treatment |
NSC34 mouse motor neurons |
[20] |
|
ALS: SOD1G93A mutated mice |
-CAPE level -Behavioural assessment - pp38 (-) |
10 mg/kg/day, orally, for 7 days after disease onset |
SOD1G93A mice |
[21] |
|
-Neuroinflammation, in vitro: 200 ng/mL LPS -in vivo, a single intraperitoneal injection of 20 mg/kg LPS, i.p., 2 h after the last CAPE injection |
-Cell viability -ERK2 -Akt -p38, -pERK1/2 -pp38 -pAKT -pJNK -EPO (+) -HO-1 (+) -iNOS (-) -COX-2 (-) -pAMPKα (+) |
-0.1 to 1.75 µM 30 min before LPS treatment, or co-treatment - 1 or 5 mg/kg once daily for 3 days |
-BV-2 murine microglial cell line -Eight-week-old male ICR mice |
[22] |
|
PD: 6-OHDA 70 µM for 6 h, on day 8–10 |
-Cell viability (+) -Cytochrome c (-) -Caspase-3 (-) -Ca2+ |
10 to 100 µM, pre-treatment for 4 h |
-Primary CGNs from 8-day-old Wistar rats -Rat liver mitochondria from 7-day-old Sprague–Dawley rats |
[23] |
|
Dopaminergic neurodegeneration: 6-OHDA, 40 μM for RMN and 70 μM for CGN |
-Free radicals (-) -Peroxynitrite (-) |
10 μM, pre-treatment for 2 h |
-Rat RMN -Primary CGNs |
[24] |
|
PD: 6-OHDA 8 mg/mL, s.i. |
-Fe, Cu, Zn and Mn (-) -ROS (-) -Protein content -TH -Mitochondrial functions: Ca2+-induced swelling, Ca2+ uptake and respiration |
-In vivo: 10 μmol/kg/day, i.p, 5 days -In vitro: 0.5 or 10 µM |
Wistar rats |
[25] |
|
-Dopaminergic neurodegeneration, in vitro: LPS/72 h -PD, in vivo: LPS 3µg/µL, intranigral, or 6-hydroxydopamine 2 µg/µL, intrastriatal, 30 minute after first CAPE injection. |
-NO (-) -ERK -p38 MAPK -HO-1 (+) -BDNF (+) -Nrf2 |
-In vitro: 3-30 µM -In vivo: 10 or 30 mg/kg/day, i.p, for 4 days |
-In vitro: rat organotypic midbrain slice cultures -In vivo: mouse model of dopaminergic neurodegeneration |
[26] |
|
PD: rotenone1 mg/kg, s.c., every 48 h, 9 injections |
-Behavioural assessment -Histopathological assessment -CD11b -COX-2 (-) -iNOS (-) -NF-κB (-) -Dopamine level (+) -TNF-α (-) -IL-1β |
2.5, 5 or 10 mg/kg/day. orally, every 48 h, 9 doses |
Male Swiss albino mice |
[27] |
|
PD: CPF 80 mg/kg, s.c. |
-PON1 activity (+) -Lipid profile -TSA (+) -TAC (+) -TOC (-) -Histopathological assessment |
10 μmol/kg/day, i.p, 21 days |
Male Swiss albino mice |
[28] |
|
PD: MPTP–HCl 20 mg/kg, i.p, four in at 2 h intervals
|
-TH-positive neurons (+) -Cell viability (+) -CAPE and MPP+ levels -DA (+) -MAO (-) -i- and nNOS (-) -Caspase-1 (-) -Cytochrome c (-) -AIF (-) -Free radicals (-) -Peroxynitrite (-) |
2, 5, or 10 mg/kg/day, 7days |
Eight-week-old male C57BL/6 mice |
[29] |
|
Loss of memory (AD): STZ 3 mg/kg, bilaterally on day 1 and 3 |
-TBARS (-) -GSH (+) -SOD (+) -CAT (+) -Nitrite (-) -AChE (-) -TNF-α (-) -eNOS (+) -NF-κB (-) -Behavioural assessment -Histopathological assessment |
6 mg/kg/day, i.p., 28 days |
Wistar rats |
[30] |
|
Dementia (AD type): STZ; 3 mg/kg, on day 1 and 3, ICV |
-MDA (-) -GSH (+) -TNF-α (-) -Behavioural tests |
3, 6 mg/kg/day, i.p, 28 days |
Wistar rats |
[31] |
|
Dementia (AD type): Aβ1-42O, unilateral stereotaxic, ICV |
-Behavioural assessment -ROS (-) -Nrf2 (+) -GSH -pGSK3α/β -Caspase-9 |
10 mg/kg/day, i.p, 1 h after brain lesion, 10 days |
Male C57Bl/6 mice |
[32] |
|
Seizures: 60 mg/kg PTZ, i.p, single dose |
-Neurological assessment -MDA (-) -NO (-) -XO -SOD (+) |
100 µmol/kg, i.p., 2 days prior to PTZ injection |
Female Swiss albino mice |
[11] |
|
Psychosis: dizocilpine maleate (MK-801), 0.5 mg/kg/day for 5 days, i.p. |
-Behavioural assessment -Histopathological assessment -MDA (-) -PC (-) -NO (-) -SOD -GSH-Px (-) -XO (-) -ADA (-) -CAT (=) |
10 μmol/kg, 6 days, started one day before MK-801, i.p.
|
Wistar rats |
[33] |
|
Diabetes: STZ 45 mg/kg, i.p, single dose |
-NO (-) -SOD -GSH-Px (-) -GSH -XO (-) -CAT (-) -MDA (-) -iNOS (-) -TNF-α (-) -IFN-γ (-) -IL-10 |
25 µM/kg/day, two days after STZ treatment for 60 days |
Male Wistar rats |
[34] |
|
Endotoxic shock: LPS, 20 mg/kg, i.p, |
-TNF-α (-) -IL-1α, -1β, -6 (-) -IL-4, -10 (+) -sICAM-1 (-) -Histopathological assessment |
10 μmol/kg/day, 14 days before shock induction and a single dose 30 min after induction |
Male Wister rats |
[35] |
|
Hepatic encephalopathy: thioacetamide:600 mg/kg, i.p., two doses (0 and 24 h) |
-Behavioural and motor assessment -Blood ammonia (=) -ALT (-) -AST (-) |
10 µmol/kg/day, i.p, starting 1 day before the first dose of thioacetamide |
Male Wistar rats |
[36] |
|
Optic nerve crushing, 10 sec |
-Apoptosis (-) -Astrocyte migration -Cell viability (+) -NF-κB (-) -IL-6 and -8 (-) -iNOS (-) -COX-2 (-) -TNF-α (-) -CCL-2 (-) |
10 μmol/kg, i.p, 10 min after the surgery |
Male Sprague Dawley rats |
[37] |
- CAPE protective effects against different neurotoxic substances
Some substances can affect the nervous system functions by damaging brain cells or nerves. Different studies investigating the protective effect of CAPE against different neurotoxic substances are described in this paragraph and summarized in Table 2.
The methotrexate (MTX) is widely used as a chemotherapeutic agent to treat various neoplastic diseases. However, it has a pronounced neurotoxicity which represents a relevant clinical problem, being directed towards the brain, the spinal cord, or the nerve roots. Its neuronal toxicity is primarily caused by demyelination as a consequence of axonal loss. MTX inhibits the conversion of folic acid to tetrahydrofolate, a molecule needed for the synthesis of DNA during S-phase and leads to improper DNA synthesis and subsequent cell death. ROS production increased indirectly by an augmented activity of purine-catabolizing enzymes such as XO and ADA. In two studies of 2006 from the same group, CAPE showed protective effects in Wistar rats treated with MTX. In the first study, CAPE reversed biochemical (ADA activity and NO levels) and histopathological parameters to the control levels in spinal cord tissue [38]. In the second study, CAPE significantly reduced MDA levels, and SOD and CAT activities in rat cerebellum, leading to protection from oxidative damage caused by MTX treatment [39]. The decreased activity of the two well-known anti-oxidant enzymes SOD ad CAT following CAPE administration could seem a contradiction. As a matter of fact, the latter could be explained by CAPE acting as non-enzymatic free radical scavengers, inhibiting oxidative stress with consequent attenuation of anti-oxidant enzyme activity. The same protection from oxidative stress was found in rat spinal cord, brainstem and sciatic nerve: CAPE treatment significantly decreased MDA level and CAT and GSH-Px activities, whereas SOD activity was increased in comparison to the MTX group [40].
Ifosfamide (IFOS) is an alkylating chemotheraputic agent used for a wide range of solid and hematologic malignancies. Commonly reported neurological complications of IFOS include acute alterations in consciousness, seizures, cerebral infarction, paralysis, neuropathies, leukoencephalopathy and ototoxicity. A study in a rat model suggested that CAPE may help in counteracting IFOS toxicity and in reducing the development of changes in the brain in clinical practice. CAPE significantly decreased MDA and PC levels, as well as caspase-3 activity, preventing neuronal apoptosis [41].
Cisplatin is another effective broad-spectrum chemotherapeutic drug. Peripheral sensory neuropathy is its major dose-limiting side effect. Recently, two studies by the same group demonstrated the protective activity of CAPE against neurotoxicity induced by cisplatin [42,43]. The first study used PC12 cells, a model of sympathetic neurons responsive to nerve growth factor (NGF). The study showed that CAPE reduced the axonal damage induced by cisplatin by promoting neuroplasticity through the activation of NGF high-affinity receptors (trkA) and the upregulation of axonal proteins related to neurite outgrowth and synaptic communication [42]. In addition, CAPE attenuated the downregulation of cytoskeleton proteins (F-actin and β-III-tubulin) and energy-related markers (AMPK α, p-AMPK α, and SIRT1) as well. Moreover, the neuroprotective mechanism of CAPE also involves the activation of the neurotrophic signaling pathways MAPK/Erk and PI3k/Akt [43].
Doxorubicin , one of the most widely used anticancer agents, is known to be neurotoxic and to induce oxidative injury and lipid peroxidation in brain. One study concluded that doxorubicin-induced oxidant injury can be prevented in rat brain by CAPE treatment, which decreased the level of MDA and increased CAT activity [44].
Cigarette smoking contains various toxic substances that can be considered harmful for the nervous tissue. CAPE was found able to have an anti-apoptotic role in the hippocampal formation of rabbits exposed to cigarette smoking. CAPE-treated rabbits showed decreased MDA level and increased SOD activity compared with untreated rabbits [45]. Similarly, brain injury in alcoholic patients can be caused by oxidative degradation of the mitochondrial genome. The combination of CAPE and intralipid treatment has a synergistic protective effect against ethanol-induced neurotoxicity: the first decreased the total oxidant status (TOS) while the intralipid treatment increased the total antioxidant status (TAS). Histopathologic results confirmed the biochemical results [46].
Acrolein is a ubiquitous pollutant existing in alcoholic beverages, water and foods such as cheese, donuts and coffee, and is also formed during combustion of organic materials, including engine exhaust, wood, tobacco and over-heated cooking oils. Acrolein-induced oxidative stress has been implicated in the pathophysiology of neurodegenerative diseases including Alzheimer’s disease. Acrolein induces hyperphosphorylation of microtubule-associated protein tau and promotes amyloid-β peptide (Aβ) aggregation in senile plaques. Pretreatment with CAPE significantly attenuated acrolein-induced neurotoxicity, ROS accumulation, and GSH depletion in HT22 mouse hippocampal cells. CAPE modulated MAPKs and Akt/GSK3β signaling pathways and restored the changes induced by acrolein in β-secretase (BACE-1) activity and/or in the activation of α-secretase (ADAM-10). These findings suggest that CAPE may provide a promising approach for the treatment of acrolein-related neurodegenerative diseases [47].
Hexavalent chromium [Cr(VI)], a proven toxin and carcinogen, is commonly used in industry and its improper disposal leads to increasing levels of Cr(VI) in water, soil and air, causing environmental pollution. Cr(VI) administration produced a significant increase in the pro-inflammatory cytokines, TNF-a and IL-6, in the rat cerebrum. Cr(VI) provoked oxidative stress and inflammation leading to activation of the JAK/STAT signaling pathway, whose dysregulation represents a key factor in neurodegenerative diseases. The neuroprotective effects displayed by CAPE could be explained by its anti-inflammatory potential. CAPE effectively decreased lipid peroxidation and NO production, and rejuvenated the altered GSH and anti-oxidant defense enzymes. In addition, it can exert a modulatory effect on the JAK2/STAT3 pathway via a SOCS3-independent mechanism. The study demonstrated that CAPE protects the brain against Cr(VI)-induced toxicity through attenuation of oxidative stress and inflammation [48]. Cadmium (Cd), a non-biodegradable heavy metal, is another environmental pollutant. Cd could cause damages to the central and peripheral neuronal systems and can induce hippocampal damage and memory deficits. A recent study showed cognitive deficits and spatial memory impairments due to CdCl2-induced neurotoxicity in mice. CAPE significantly decreased CdCl2-induced neuronal apoptosis and the expression of Bax and cleaved caspase-3, while promoting Bcl-2 expression in mice hippocampus. CAPE also inhibited the CdCl2- initiated Aβ accumulation and the activation of pro-inflammatory factors and of microglia in the brain [49].
Neurotoxicity can be also caused by poisoning with organophosphate pesticides, which results in cholinergic syndrome. Seeking an alternative or supportive treatment for organophosphate insecticide poisoning, a study investigated the effect of CAPE on chloropyriphos toxicity. Although CAPE had a significant effect on protection of neuronal degeneration by decreasing the amount of oxidative stress (TOS), results revealed that CAPE significantly inhibits the enzyme AChE. Consequently, it results not suitable for the management of organophosphate toxicity [50], where the acetilcholinesterase activity results already compromised
In order to decrease the risk of drug resistance, a commonly used strategy is the combination of various drugs to treat a disease. Isoniazid (INH), a first line drug for tubercolosis, has side effects on both central and peripheral nervous system while optic neuropathy has been reported with the administration of ethambutol (ETM). Tuberculosis treatment can be discontinuous because of such neurotoxic side effects. Interestingly, one study demonstrated that CAPE can protect against INH- and ETM-induced neurotoxicity in rat brain and sciatic nerve. CAPE normalized the levels of both MDA and TOS, increased by anti-tuberculosis medications, and prevented the down-regulation of SOD and PON-1 activities, by scavenging free radicals produced following INH and ETM administration. CAPE treatment reduced effectively the edema and vascular congestion caused by these medications in both central and peripheral nervous systems [51].
Sevoflurane, a volatile anesthetic frequently used for pediatric anesthesia, was reported to promote neurodegeneration. A recent study demonstrated that CAPE effectively inhibited sevoflurane‑induced neuroapoptosis in rat pups, by modulating the expression and phosphorylation of apoptotic proteins, MAPKs, and of the PI3K/Akt pathway. CAPE significantly reduced sevoflurane‑induced apoptosis, down-regulated the expression levels of caspases and pro‑apoptotic proteins (Bax and Bad) and elevated the expression levels of Bcl‑2 and Bcl‑xL, when compared with sevoflurane treatment [52].
Table 2: CAPE protective effects against different neurotoxic substances. Where applicable the effect of CAPE on the parameters measured is reported between parenthesis (+ or -).
|
Neurotoxic substance |
Parameters measured |
CAPE dose |
Animal/Cell used |
Ref. |
|
MTX: 20 mg/kg, i.p., single dose |
-Histopathological assessment -ADA (-) -NO (-) |
10 µmol/kg/day, i.p, for 7 days |
Male rats |
[38] |
|
MTX: 20 mg/kg, i.p., single dose on day 2 |
-MDA (-) -SOD (-) -CAT (-) |
10 µmol/kg/day, i.p, for 7 days |
Male rats |
[39] |
|
MTX: 20 mg/kg, i.p., single dose on day 2. |
-MDA (-) -SOD (+) -GSH-Px (-) -CAT (-) |
10 μmol/kg/day, i.p., for 7 days |
Wistar male rats |
[40] |
|
IFOS: 300 and 500 mg/kg, i.p., two doses |
-Carbonyl content (-) -CAT -MDA (-) -Caspase-3 (-) |
10 µmol/kg/days, i.p, for 2 days, starting 1 day before injection of IFOS |
Wistar male rats |
[41] |
|
Cisplatin: 5and 32 μM |
-Cell viability (+) -Neurite outgrowth (+) -GAP-43 (+) -Synapsin I (+) -Synaptophysin (+) |
1, 5, 10, 25, 50, and 100 μM for 24 h |
-PC12 cells -SH-SY5Y cells |
[42] |
|
Cisplatin: 5 μM |
-Protein content (+) -Glucose uptake (+) -Glutamate uptake -ROS (-) -F-Actin (+) -β-III-Tubulin (+) -SIRT 1 (+) -AMPK α and pAMPK α (+) |
10 μM |
-PC12 cells -transfected COS-7 cells -transfected HEK cells -glial cells |
[43] |
|
Doxorubicin: 20 mg/kg i.p., single dose |
-MDA (-) -NO (-) -GSH-Px -CAT (+) -SOD |
10 μmol/kg/day, i.p, for 12 days starting 2 days before doxorubicin |
Male Sprague Dawley rats |
[44] |
|
Cigarette smoke: 1 hour daily for 4 weeks |
-MDA (-) -SOD (+) -Apoptosis (-) |
10 mmol/kg/day, i.p, for 4 weeks before the exposure to cigarette smoke |
Rabbits |
[45] |
|
Ethanol: 3 mg/kg, oral |
-TOS (-) -TAS (=) -Histopathological assessment |
10 μmol/kg, i.p., immediately after ehanol administration |
Rats |
[46] |
|
Acrolein: 1 M |
-Cell viability (+) -ROS (-) -GSH (+) -MAPKs -Akt/GSK3 -α/β-secretase |
0-90 μM, pretreatment for 30 min |
HT22 mouse hippocampal cells |
[47] |
|
K2CrO4: 2 mg/kg/day, i.p., for 30 days |
-SOCS3, JAK2 and STAT3 -NO (-) -GSH (+) -SOD (+) -GSH-Px -AChE -TNF-α (-) -IL-6 (-) |
20 mg/kg/day, orally, for 30 days |
Wistar male rats |
[48] |
|
CdCl2: 1.5 mg/kg |
-Neurobehavioural assessment -Histopathological assessment -AMPK and pAMPK -SIRT1 -Bcl-2 (+) -Bax (-) -Caspase-3 (-) -p-Tau -TLR4 -IL-6 (-) -IL1-β (-) -TNF-α (-) |
10 μmol/kg/ day, for 4 weeks |
7 weeks old Kunming mice |
[49] |
|
Chlorpyriphos: 10 mg/kg, oral |
-AChE (-) -TOS (-) -TAR -Histopathological assessment -Caspase-3 -Bcl-2 -Bax |
10 μmol/kg, i.p., immediately after chlorpyriphos admnistration |
Wistar rats |
[50] |
|
INH and ETM: 50 mg/kg/day, orally, for 30 days |
-Histopathological assessment -MDA (-) -TOS (-) -TAC (+) -SOD (+) -PON-1 (+) -NO (-) |
10 mol/kg/day, i.p., for 30 days |
Male Sprague-Dawley rats |
[51] |
|
Sevoflurane: (2.9%) for 6 h at day 7 |
-Caspase‑3, -8 and -9 (-) -Bax (-) -Bcl‑2 (+) -Bcl‑xL (+) -Bad (-) -MAPK (-) -JNK -ERK -PI3K (-) |
10, 20 or 40 mg/kg, from postnatal day 1 to day15 |
Rat pups |
[52] |
- CAPE protective effects against ischaemia
Approximately 87% of all brain strokes are ischaemic ones. Ischeamic stroke is caused by a blockage in an artery that supplies blood to the brain. The blockage reduces the flow of blood and oxygen to the brain, leading to damage or death of brain cells. If circulation is not restored quickly, brain damage can be permanent. Ischaemic brain damage is an evolving process which begins during the insult and extends into the recovery period after the reperfusion interval. The immediate area surrounding the infarct consists of neurons undergoing either necrosis or apoptosis.
Many studies investigated the potential protective role of CAPE in various CNS ischaemic conditions (Tab. 3). In one study the effects of CAPE and alpha-tocopherol, another free radical scavenger, on ischaemia–reperfusion cerebral injury were compared. CAPE proved to be better than alpha-tocopherol in reducing the cerebral level of MDA, a marker of oxidative stress [53]. CAPE was also found able to significantly reduce the total infarct volume following focal cerebral ischaemia. Its effects were found related to its antioxidant activity and to the up-regulation of NO production [54], which has many beneficial properties in ischemia–reperfusion injury, including increase of blood flow produced by cerebral vasodilation. Interestingly, Khan et al. showed that the protective effect of CAPE after transient focal cerebral ischaemia and following reperfusion is achieved through not only anti-oxidant and anti-inflammatory mechanisms, but also via hemodynamic effects in a dose-dependent manner, in both short- and long-term ischaemia/reperfusion models. CAPE increased nitric oxide and glutathione levels, decreased lipid peroxidation and nitrotyrosine levels, and enhanced cerebral blood flow. A down-regulation of the inflammation by blocking of the NF-κB activity was also observed. The affected mediators included adhesion molecules (ICAM-1 and E-selectin), cytokines (TNF-α and IL-1β) and inducible nitric oxide synthase. This action was further confirmed by the reduction of the expression of ED1, a marker of activated macrophage/microglia. CAPE also inhibited apoptotic cell death, by down-regulating caspase-3 and upregulating the anti-apoptotic protein Bcl-xL [55]. These results were supported by a study on rabbits [56], in which CAPE attenuated the elevation of plasma level of MDA, CAT and XO caused by cerebral ischaemia injury and restored the plasma level of GSH and NO. In the same in vivo model, CAPE significantly attenuated serum S-100B level, an index for brain damage, after middle cerebral artery occlusion [57]. As for the timing of CAPE administration, Cengiz et al. [58] showed that the pre-treatment reduced neuroglia activation and structural changes (infarcted areas, pyknotic cells, vacuolization and neuroglial cell infiltration) in rat cerebral cortex after middle artery cerebral ischaemia/reperfusion. On the other hand, post-treatment of ischaemic brain injury with CAPE significantly reduced the infarct size, the IL-1α production, and the expression, in a dose-dependent manner, of TNF-α, the hypoxia inducing factor (HIF)-1α, the monocyte chemoattractant protein (MCP)-1, and the indoleamine 2,3-dioxegenase (IDO) in the cerebral cortex ipsilateral to the photothrombosis. In addition, CAPE elicited a significant increase in HO-1 expression and IL-10 production, demonstrating that its remarkable neuroprotective effect on ischaemic brain injury is exterted via its anti-inflammatory properties [59]. Feng and collaborators reported in 2008 [60] that CAPE compensated the functional alterations in mitochondria isolated from mouse brain challenged by anoxia-reoxygenation. This effect was attributed to the inhibition of the decrease in membranes fluidity and of lipoperoxidation and protein carbonylation. A blockade of the enhanced release of cardiolipin and cytochrome c was also observed. Interestingly CAPE was used at concentrations ranging from pico- to micromolar, making CAPE more efficient in quenching free radicals than its derivatives, ferulic acid and ethyl ferulate.
The effect on isolated brain mitochondria was confirmed in another paper where CAPE directly inhibits Ca2 +-induced cytochrome c release. In the same study CAPE proved able to prevent neuronal death in neonatal rats exposed to hypoxia–ischaemia. This condition is associated with clinical syndromes of neurological disability, such as seizures, intellectual impairment and cerebral palsy. CAPE can effectively protect against neuronal and tissue loss in the cortex, hippocampus and thalamus in vivo. In addition, CAPE inhibited hypoxia-induced caspase-3 activation, hypoxia-mediated expression of inducible nitric oxide synthase and caspase-1 in vivo. It also potently blocked nitric oxide-induced neurotoxicity in vitro [61].
In another model of stroke CAPE was tested on the subarachnoid hemorrhage (SAH) and was found able to significantly attenuate the brain ischaemia due to vasoconstriction after SAH: CAPE inhibited the marked narrowing in the lumens and the thickening in the walls of basilar arteries, as well. The tissue level of MDA were also found decreased after CAPE administration following SAH, because of ROS scavenging, lipid peroxidation prevention and GSH and NO increase in cerebral tissue [62]. In a similar model, CAPE was shown to reduce hippocampal neuron loss induced by SAH, but no significant effect of CAPE on vasospasm was found [63].
Injury to the gray matter of the spinal cord, with consequent neuronal death, has generally been considered an important element in the pathology of spinal cord ischaemic injury. The latter represents the main complication in surgical repair of thoracic and thoraco-abdominal aneurisms and remains a persistent clinical problem. The most serious complication is paraplegia. Methylprednisolone (MP) proved effective in improving neurologic function after traumatic spinal cord injury. Ilhan et al [64] compared the effects of CAPE and MP on histopathological changes, antioxidant status, lipid peroxidation, and neurologic recovery in temporary induced spinal cord ischaemia in rabbits. CAPE reduced ischaemic and reperfusion damage and provided better neurologic outcome than MP. CAPE administration resulted in an improvement in microcirculatory environment during reperfusion, preventing endothelial cell lysis provoked by proteases released by activated leukocytes.
Table 3: CAPE protective effects against ischaemia/reperfusion injury. Where applicable the effect of CAPE on the parameters measured is reported between parenthesis (+ or -).
|
Stimulus |
Parameters measured |
CAPE dose |
Animal used |
Ref. |
|
Bilateral CCA occlusion (20 min) then reperfusion (20 min)
|
-ADA -XO -SOD -GSH-Px -CAT -NO -MDA (-) |
10 µmol/kg, i.p. 10 min. after placing the occlusive vascular clamps |
Sprague–Dawley rats |
[53] |
|
Cerebral infarction: right MCA occlusion and bilateral CCA clipping, 60 min |
-NO (+) -Histopathological assement |
0.01, 0.1, 1 and 10 µg/kg, i.v. 15 min before MCA occlusion |
Male Long–Evans rats |
[54] |
|
MCA occlusion, 20 or 90 min |
-Histopathological assessment -Neurological assessment -TNF-α (-) -IL-1β (-) -iNOS (-) -ED1 (-) -Bcl-xL (+) -Caspase-3 (-) -NO (+) -TBARS -GSH (+) -NF-κB (-) -MDA (-) -ICAM-1 (-) -E-selectin (-) -Nitrotyrosine (-) |
1–10 mg/kg, i.v., either at or after reperfusion |
Male Sprague–Dawley rats |
[55] |
|
Right permanent MCA occlusion |
-Histopathological assessment -Neurological assessment -MDA (-) -GSH (+) -CAT (-) -NO (+) -XO (-) |
10 µmol/kg/day, i.p., after occlusion for 7 days |
Male New Zealand rabbits |
[56] |
|
Permanent MCA occlusion |
-Serum S-100B (-) |
10 µg/kg/day, i.p. for 7 days after occlusion |
Male New Zealand rabbits |
[57] |
|
MCA occlusion (60 min), followed by 24 h reperfusion |
-Structural changes |
50 µM/kg, i.p., once before occlusion |
Wistar rats |
[58] |
|
Cortical ischaemia: skull irradiation with cold light laser in combination with systemic administration of rose bengal |
-Histopathological assessment -TNF-α (-) -HIF-1α (-) -MCP-1 (-) -IDO (-) -HO-1(+) -IL-1α (-) -IL-10 (+) |
0.5-5 mg/kg, i.p., 1 and 6 h after ischaemic insult |
Male C57BL/6 mice |
[59] |
|
Anoxia-reoxygenation |
-Mitochondrial oxygen consumption -Mitochondrial anisotropy -Mitochondrial TBARS -Mitochondrial protein concentrations -Protein carbonylation (-) -CL and Cytochrome c release (-) |
10-10−5 μM before the anoxia or just at reoxygenation |
Male Kunming mice |
[60] |
|
CCA ligation followed by exposure to hypoxia |
-i- and nNOS (-) -Cytochrome c (-) -Caspase-1 and -3 (-) |
40 mg/kg/day, 4 hrs before and/or after the stimulus |
7-day-old Sprague–Dawley rats |
[61] |
|
SAH |
-MDA (-) -GSH (+) -NO (+) -Histopathological assessment |
10 µmol/kg, i.p. twice daily for 5 days after SAH |
15-week-old male Wistar rats |
[62] |
|
SAH |
-Histopathological assessment |
10 mg/kg/day, twice a day, for 3 days starting 6 h after SAH. |
Wistar rats |
[63] |
|
Aortic occlusion, 21 min |
-MDA (-) -SOD -CAT -Histopathological assessment -Neurologic assessment |
10 µmol/kg, i.p. 30 min before the stimulus |
New Zealand rabbits |
[64] |
- CAPE protective effects against injury
The secondary injury consists of the biochemical and histopathological cascade mechanisms in brain and spinal cord following a primary injury and leading to progressive neuronal death after trauma. Table 4 lists the studies investigating the protective effect of CAPE against these mechanisms.
A clip compression model was used in rats to simulate the spinal cord injury (SCI). In the early phase of injury, CAPE suppressed the serum levels of pro-inflammatory cytokines, i.e. TNF-α and IL-1β. At the same time, CAPE decreased hemorrhage and necrosis occurrence both in gray and white matter, as well as in the central canal as demonstrated by the histopathological evaluation of the healing process [65]. Another study compared the effects of CAPE with those of methylprednisolone (MP) in the prevention of neurological deficits caused by secondary spinal cord injury. CAPE has been found to be superior to MP in preventing apoptosis without showing the latter side-effects, namely gastrointestinal hemorrhage and infection [66]. The effects of intrathecal CAPE administration following SCI were investigated as an alternative approach in CNS diseases, because the systemic administration of pharmacological agents does not guarantee the attainment of the maximum effective dose in the damaged area. The intrathecal CAPE administration resulted in a decrease in tissue and serum levels of IL-6, an inflammatory cytokine that plays an essential role in secondary damage. Although no significant decrease was identified in TNF-α level, CAPE led to a decrease in the edema and microhemorrhage that developed in association with a local inflammatory response in the damaged area. In addition, CAPE was more effective than MP in decreasing microhemorrhage [67]. Interestingly, the intratechal administration of CAPE and MP decreased also the MDA, TOA, and SOD levels, with a greater effect in the CAPE group [68]. Since CAPE and MP treatment substantially increased TAC levels, it was concluded that intrathecal injection of both compounds can inhibit the lipid peroxidation and the oxidant increase following SCI. In a model of SCI caused by hemi-transection, CAPE suppressed the expression of mRNA of the pro-inflammatory cytokine IL-1β and of the two inflammatory enzymes iNOS and COX-2. An enhanced recovery of locomotor function and a reduction of the lesion size were also found following CAPE treatment [69].
One of the early pathological events triggered by traumatic brain injury (TBI) is the breakdown of the blood brain barrier (BBB), which leads to the infiltration of fluid and circulating cells and molecules, resulting in an exacerbation of brain damage and tissue loss. Recently, myeloperoxidase (MPO) analysis was used to identify the level of polymorphonuclear leukocytes (PMNL) infiltration after TBI. PMNL worsen brain damage due to the release of cytotoxic mediators that interfere with BBB function. Hypochlorite acid is formed through the MPO-H2O2–Cl-system and is one of the oxidants released by activated leukocytes. CAPE administration to mice with TBI showed a significant decrease in MPO levels thereby inhibiting the BBB inflammatory process [70]. Another study examined the potential role of CAPE in reducing some of the pathological consequences of TBI in rodent models. Post-TBI administration of CAPE reduced BBB permeability and cortical tissue loss both in rats and mice. The improvement was associated with the preservation of the levels of the tight junction protein claudin-5. On the other hand, it failed to offer significant improvements in either motor or memory tasks [71]. The administration of a single dose of CAPE post-TBI can also have protective effects via decreasing the elevated MDA levels and restoring the activities of antioxidant enzymes (SOD and GSH-Px), with the exception of CAT. The morphology of neurons was well preserved, and the immunoreactivity of degenerating neurons after trauma was reduced in the CAPE treated group [72].
Since TBI has a multi-mechanistic etiology, a recent study combined the endogenous nitric oxide (NO) metabolite S-nitrosoglutathione (GSNO) with CAPE to accelerate the rate and enhance the degree of recovery. GSNO is a natural component of the human body, present in the brain and other organs. It provides neuroprotection and improves neurobehavioural functions via anti-inflammatory and anti-neurodegenerative mechanisms. It acts via S-nitrosylation of target proteins, including NF-κB, STAT3, COX-2, caspase-3, calpains, and i- and eNOS. The combination therapy improved cognitive and depressive-like behaviour compared with GSNO or CAPE monotherapy. Separately, both GSNO and CAPE improved mitochondrial integrity/function and decreased oxidative damage; however, the combined therapy had greater effects on dynamin-1-like protein (Drp1) and manganese-dependent superoxide dismutase (MnSOD). Additionally, while CAPE alone activated 5'-adenosine monophosphate-activated protein kinase (AMPK), this activation was heightened in combination with GSNO. These results recommended that the combination therapy-based multi-mechanistic approach is worthy of investigation in human TBI, because none adverse effect for both GSNO and CAPE is known [73].
Table 4: Protective effects of CAPE against injury. Where applicable the effect of CAPE on the parameters measured is reported between parenthesis (+ or -).
|
Stimulus/injury |
Parameters measured |
CAPE dose |
Animal/Cell used |
Ref. |
|
SCI: aneurysm clip |
-IL-1β (-) -TNF-α (-) -Histopathological parameters |
10 μg/kg, i.p, 30 min after trauma |
Male Wistar rats |
[65] |
|
Paraplegia: epidural clip application for 60 sec. |
Histopathological parameters |
10 μmol/kg, i.p |
Female Sprague-Dawley rats |
[66] |
|
SCI: Yasargil aneurysm clips |
-IL-6 (-) -TNF-α (=) -Histopathological parameters |
1 μg/kg, following SCI induction |
Female Wistar rats |
[67] |
|
SCI: Yasargil aneurysm |
-MDA (-) -TOA (-) -TAC (+) -SOD (-) -GSH-Px |
1μg/kg, single dose |
Female Wistar rats |
[68] |
|
SCI: hemitransection |
-Locomotor function -Histopathological parameters -IL-1β (-) -iNOS (-) -COX-2 (-) |
2 or 10 μmol/kg/day, i.p, for 28 days |
Female Wistar rats |
[69] |
|
Head trauma with marmarou model |
MPO activity (-) |
10 mg/kg, i.p, 24 h before trauma and 30 min after trauma and every day for 7 days |
Male Sprague mice |
[70] |
|
Brain trauma: CCI injury model |
-Blood–brain barrier (BBB) integrity (+) -Claudin-5 expression (+) -Neurobehavioural assessment |
10 mg/kg, i.p, -30 min following injury and/or daily for the next 4 days |
-Male Sprague-Dawley rats -C57BL/6 mice |
[71] |
|
TBI: using cranial impact to the skull from a height of 7 cm at a point just in front of the coronal suture and over the right hemisphere. |
-MDA (-) -SOD (+) -GSH-Px (+) -CAT (=) -Histological examinations -Caspase- 3 |
10 μmol /kg/i.p, single dose15 min after trauma |
Male Sprague–Dawley rats |
[72] |
|
TBI: focal CCI technique |
-Neurobehavioural parameters -Histopathological parameters -AMPK and pAMPK (+) -Fission (Drp1 and Fis 1) and fusion (Opa1)-associated proteins (+) -Mitochondrial factor PGC1α -HO-1 -MnSOD (+) |
5 mg/kg, plus GSNO 0.05 mg/kg, 2 h after CCI, i.v. and then daily orally |
Young adult male wild type C57BL/6 mice |
[73] |
- CAPE anti-tumoral effects in CNS
In 2020 Cancer Stat Facts, an estimated 23,890 new cases in USAwill be diagnosed with primary cancerous tumors of the brain and spinal cord. The 5-year survival rate for people with a CNS tumor is around 32.6% [74]. The most common types of brain tumors are gliomas and meningiomas. Gliomas are considered the most malignant form of brain tumors, and are ranked among the most aggressive human cancers. Choi et al in 2007 [75] demonstrated that the anti-oxidant property of CAPE can exert a pro-apoptotic effect in Fas-mediated cell death in human malignant astrocytoma cells. Fas activation increased intracellular ROS levels in a NADPH oxidase- and caspase-dependent manner. Choi's results suggested that Fas-induced ROS production protects astrocytoma CRT-MG cells from caspase-dependent apoptosis. Suppression of Fas-induced ROS levels augments Fas-mediated apoptosis by enhancing the enzymatic activity of caspase-3. CAPE, by inhibiting ROS production, dramatically sensitized astrocytoma cells to Fas-induced loss of mitochondrial transmembrane potential and subsequent cell death. Another study demonstrated that CAPE suppressed invasion and proliferation of glioma cells by downregulating of phospholipase D1 (PLD1) expression at the transcriptional level. PLD is a class of enzymes functionally linked with oncogenic signals and tumorigenesis. CAPE exerted its effect via inhibition of binding of NF-κB to PLD1 promoter. In addition, CAPE bound to a Cys837 residue of the protein PLD1 and inhibited its enzymatic activity. The activation of matrix metalloproteinases-2 induced by phosphatidic acid, a product of PLD activity, was also inhibited [76].
5-lipoxygenase (5-LO) is another enzyme involved in the pathophysiology of glioma. Strong 5-LO expression has been observed in glioma cells and inhibiting this enzyme could impact glioma cell proliferation. Interestingly, CAPE (1 µM) was found effective in reducing 5-LO activity more efficiently than the ferulic acid ester (FAPE) in two human glioma cell lines, Hs683 and LN319. CAPE administration led to cytotoxicity, cell growth impairment and apoptosis occurrence [77].
In an in vivo model, Bio 30, a water-miscible CAPE-rich extract of New Zealand propolis, was analyzed for its anti-tumoral properties. Bio 30 suppressed completely the growth of both neurofibromatosis type1 (NF1) and neurofibromatosis type 2 (NF2) tumor xenografts in mice, suggesting that CAPE-rich propolis might serve as an effective, inexpensive and safe NF therapeutic first approach. Seventy percent of human cancers and all NF tumors, but not normal cells, are highly dependent on PAK1 for their growth. PAK1 is a Rac/CDC42-dependent Ser/Thr kinase. Evidence was found that CAPE, by blocking the oncogenic PAK1 signaling pathways, is the anti-tumor ingredient of Bio 30 [78]. All the studies described above about the anti-tumor effect of CAPE in CNS are listed in table (5).
Table 5: CAPE anti-tumoral effects in CNS. Where applicable the effect of CAPE on the parameters measured is reported between parenthesis (+ or -).
|
Stimulus/tumor |
Parameters measured |
CAPE dose |
Ref. |
|
Human astrocytoma (CRT-MG cells) |
-Cell viability (-) -ROS (-) -Caspase-3 and -8 (+) -NOX4 -DEVDase activity |
0-25 µg/ml pre-treatment for 1 h |
[75] |
|
Malignant brain tumor: human U87MG glioma cells |
-PLD (-) -PLD1 (-) -PLD2 -NF-κB-binding motif (-) -α-tubulin -MMP-2 (-) -Invasion assay (-) -Gelatin zymography |
10, 20 µM for 24 h |
[76] |
|
-HEK293 cells stably co-transfected with a pcDNA3.1 vector expressing 5-LO and a pBUDCE4.1 vector expressing 5-LO activating protein -Human glioma cells: Hs683 and LN319. |
-Cell viability (-) -Molecular Docking -5-LO activity (-) |
-1 μM, pre-incubation -10 µM, for 3 days |
[77] |
|
Tumor xenografts in nu/nu mice: SC injection of NF1-deficient MPNST (S-462) cells or NF2-deficient Schwannoma (HEI-193) cells |
-Cell viability (-) -Tumor size (-)
|
Bio 30 (a CAPE-rich extract), 100–300 mg/kg, i.p., twice a week |
[78] |
- CAPE derivatives
Due to the involvement of a variety of pathways in neurological disorders, the finding of molecules with multiple neuro-protective effects would be a great achievement for the slowing down of the course of these diseases. CAPE is supposed to be a promising neuroprotective agent with multiple targets. Many studies aimed to explore the structure-activity relationship of CAPE in order to optimize its structure through a series of studies on CAPE derivatives. Figure 2 shows the chemical structures of CAPE and of some of its analogues. Novel derivatives of CAPE were recently designed, synthesized and evaluated in different in vitro models as potential neuroprotective agents. Phenolic hydroxyl groups and double bonds in the structure of CAPE proved critical in conferring neuroprotective properties to the compound, whereas monophenol compounds showed low anti-inflammatory activity. Compounds with a phenolic hydroxyl group on para-position exerted a higher anti-inflammatory activity than compounds with the same group on ortho-position, indicating that conjugated double bond played an important role in anti-inflammatory activity. Only compounds with retained catechol structure showed anti-oxidant properties. Ami4 group did not show the same anti-oxidant activity found for the bioisoteric group hydroxyl group. Replacing the aromatic alkyl part of CAPE with different liposoluble groups improved the anti-inflammatory and anti-oxidant activities and the BBB permeability [79], measured with parallel artificial membrane permeability assay (PAMPA), a high throughput technique developed to predict passive permeability through biological membranes. These findings were supported with a study on PC12 cells suggesting that the presence of the caffeic acid group is important to keep the cytoprotective and neuritogenic activities of the compound [80]. Another group explored neuroprotective effects of CAPE and another two caffeic acid derivatives, Danshensu and Curcumin. They are similar in structure, since they have an aromatic ring, but they differ in the numbers of hydroxyl groups attached to the aromatic ring and in the conjugated double bond. The structure-neuroprotective activity relationship against H2O2-induced oxidative damage in PC12 cells and the D-galactose (D-gal)- induced cognitive impairment was investigated in mice. The neuroprotection exterted by these compounds was mediated by the activation of the protein kinase A (PKA)-cyclic AMP response element-binding protein (CREB) pathway. It was found that 3, 4-dihydroxyphenyl hydroxyl groups attached to the aromatic ring play a more important role than conjugated double bond in up-regulating the expression of PKA/CREB pathway in the mouse hippocampus [81].
In two separate studies, CAPE proved active in suppressing invasion and proliferation of glioma cells by downregulation of PLD1 and 5-LO expression. One study showed that CAPE analogues with a catechol moiety, dihydroxydihydrocinnamic acid phenethylester (DHHC) and dimethoxycinnamic acid phenethyl ester (DMC) inhibit expression of PLD1 in a lower potency than that of CAPE due to differences in NF-κB transactivation. This was confirmed by structural analysis, which showed that both electrophiles in CAPE, Michael reaction acceptor and catechol moiety, are involved in the downregulation of PLD1 expression and in NF-κB transactivation [76]. The other study demonstrated that CAPE, but not FAPE, displayed substantial cytotoxicity against two glioma cell lines [77].
Even though CAPE stability in human plasma was demonstrated [9], its lipophilic properties, due to long carbon groups in an aromatic and aliphatic structure, can hamper CAPE absorption upon oral administration. Hence, FA-97 caffeic acid phenethyl ester 4-O-glucoside was synthesized to overcome low water solubility and poor bioavailability of CAPE, which represent the main limits to its application in vivo. The in vivo and in vitro results showed that FA-97 is able to suppress oxidative stress-mediated neuronal cell apoptosis, and to have protective properties against scopolamine-induced cognitive impairment. Since the pathway involved in such activity is the Nrf2/HO-1 signaling, FA-97 can be suggested to have a therapeutic potential in AD [82]. The low bioavailability of CAPE could also be ascribed to rapid decomposition by esterases. Another study aimed to synthesize CAPE analogs with better stability and higher BBB permeability, found in various in vitro models that E-3,4-dihydroxy styryl sulfonamides and their 3,4-diacetylated derivatives have better neuroprotective activities against oxidative and inflammatory injury than CAPE. The same compounds showed high blood–brain barrier permeability [83] predicted by PAMPA test. KS370G, a novel caffeic acid phenylethyl amide significantly inhibited neuroinflammation in CNS by inhibition of the NO release and of the iNOS and COX-2 expression. KS370G treatment also induced HO-1 activity and suppressed cytokine signaling (SOCS)-3 expression in the microglia [84].
Centrally active catechol O-methyltransferase (COMT) inhibitors can have a role in the treatment of dopamine deficiency-related neurological disorders such as PD, depression and schizophrenia. First generation COMT inhibitors, like tolcapone and entacapone, are based on simple catechol scaffolds and their clinical utility was hampered due hepatotoxicity in addition to poor bioavailability. Recently, new nitrocatechol COMT inhibitors (Fig. 2) based on naturally-occurring CAPE were developed. Their effect was within the nanomolar range with lower hepatotoxicity in addition to a good BBB permeability [85] evaluated by PAMPA assay. Lead optimization efforts opened a new window on repurposing of nitrocatechols beyond their established role as COMT inhibitors. The results of another study showed that the nitrocatechol scaffold is required for a significant inhibition of hyperphosphoryled tau protein aggregation. Tau hyperphosphorylation and assembly into intracellular neurofibrillary tangles triggers neurodegeneration and was largely observed in AD. The activity of these compounds was enhanced by introducing bulky substituents at the side chain and by αcyanocarboxamide derivatization as amide bond provides superior conformational stability [86].
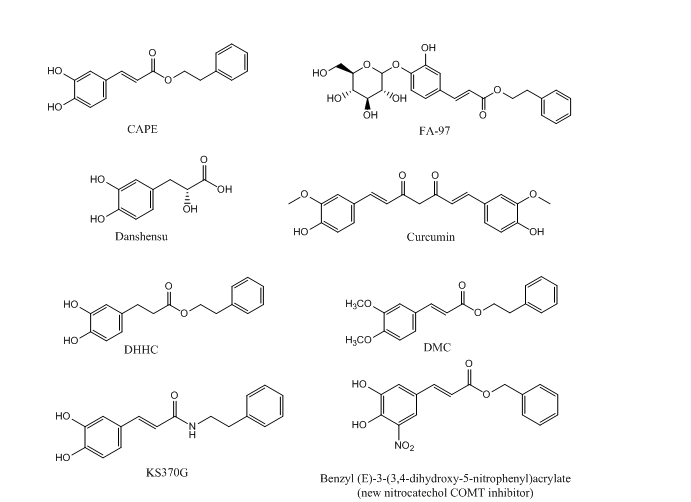
Figure 2. Chemical structure of CAPE and CAPE analogues
- Conclusions
CAPE, the most investigated compound of propolis, has a wide range of pharmacological activities. It has strong neuroprotective activity and its effects have been studied in several pathological conditions affecting the CNS. The main goal of this review is to provide a comprehensive view into the role of CAPE in the management of these disorders by focusing on the published data about CAPE and neuroprotective effects. In this review also we shed the light on its molecular mechanisms of action. In addition it summarizes the structure activity relationship of CAPE synthetic derivatives which will be helpful for further development of neuroprotective derivatives.
Abbreviations
5-LO: 5-lipoxygenase
6-OHDA: 6-Hydroxydopamine
AChE: acetylcholinesterase
AD: Alzheimer’s disease
ADA: adenosine deaminase
AIF: Apoptosis-inducing factor
ALS: Amyotrophic lateral sclerosis
ALT: alanine transaminase
AST: aspartate transaminase
AMN: adrenomyeloneuropathy
AMPK: 5'-adenosine monophosphate-activated protein kinase
Bax: Bcl‑2‑associated X protein
Bad: Bcl‑2‑associated agonist of cell death
BBB: blood brain barrier
Bcl‑2: B cell CCL/lymphoma 2
Bcl‑xL: Bcl‑2‑like 1
BDNF: brain-derived neurotrophic factor
CAPE: caffeic acid phenethyl ester
CAT: Catalase
CCA: common carotid arteries
CCI: controlled cortical impact
CCL‑ 2: C‑C.motif ligand-2
CGNs: cerebellar granule neurons
CNS: central nervous system
COMT: catechol O-methyltransferase
COX-2: cyclooxygenase-2
CPF: Clorpyrifos-ethyl
CREB: cAMP-responsive element binding protein
DA: dopamine
DCF: dichlorofluorescein assayDHHC: dihydroxydihydrocinnamic acid phenethylester
DMC: dimethoxycinnamic acid phenethyl ester
EAE: experimental autoimmune encephalomyelitis
ED1: marker of activated macrophage/microglia
eNOS: endothelial nitric oxide synthase
ERK: extracellular signal‑regulated kinase
EPO: erythropoietin
ETM: ethambutol
FAPE: ferulic acid esterGSH: glutathione
GSH-Px: glutathione peroxidase
GSNO: S-nitrosoglutathione
HE: encephalopathy
HIF-1α: hypoxia inducing factor-1α
HO-1: heme oxygenase
ICAM-1: Intercellular adhesion molecule-1
ICV: intracerebroventricular
IDO: indoleamine 2,3-dioxegenase
IFN- γ: interferon-γ
IFOS: ifosfamide
IκB: inhibitor κB
IKK: inhibitor of nuclear factor-κB (IκB) kinase
IL-1β: interleukine-1β
INH: isoniazid
iNOS: inducible nitric oxide synthase
JNK: c‑Jun N‑terminal kinase
LPS: lipopolysaccharide
MAO: monoamine oxidase
MAPK: mitogen‑activated protein kinases
MBP: Myelin Basic Protein
MCA: middle cerebral artery
MCP-1: monocyte chemoattractant protein-1
MDA: Malondialdehyde
MK-801: dizocilipine maleate
MKK4: Mitogen-Activated Protein Kinase Kinase 4
MMP: matrix metalloproteinase
MP: Methylprednisolone
MPO: myeloperoxidase
MPP+: 1-methyl-4-phenylpyridinium
MPTP: methyl-4-phenyl-1,2,3,6-tetrahydropyridine
MS: multiple sclerosis
MTX: methotrexate
NF: neurofibromatosis
NF-κB: nuclear factor-κB
NGF: nerve growth factor
NMDA: N-methyl-D-aspartate
NO: nitric oxide
Nrf2: nuclear factor erythroid 2-related factor 2
PAMPA: parallel artificial membrane permeability assay (PAMPA)PC: protein carbonyl
PD: Parkinson’s disease
PFC: prefrontal cortex
PI3K: phosphoinositide 3‑kinase
PLD1: phospholipase D1
PMNL: polymorphonuclear leukocytes
PON1: paraoxonase
PTZ: pentylenetetrazole
RMN: rostral mesencephalic neurons
ROS: reactive oxygen species
SAH: Subarachnoid hemorrhage
SCI: spinal cord injury
SE: status epilepticus
SOD: superoxide dismutase
STZ: streptozotocin
TAC: total antioxidant capacity
TAK1: transforming growth factor-β-activated kinase 1
TAR: total antioxidant response
TAS: total antioxidant status
TBARS: thiobarbituric acid reactive substances
TBI: traumatic brain injury
TH: tyrosine hydroxylase
TNF‑α: tumor necrosis factor-α
TOA: total oxidant activity
TOC: total oxidant capacity
TOS: Total oxidant status
TRAF2: TNF Receptor Associated Factor 2
TSA: total sialic acid
VLCFA: very long chain fatty acids
X-ALD: X-linked adrenoleukodystrophy
XO: xanthine oxidase
References
- Menezes da Silveira, C.C.; Luz, D.A.; da Silva, C.C.; Prediger, R.D.; Martins, M.D.; Martins, M.A.; Fontes‐Júnior, E.A.; Maia, C.S. Propolis: A useful agent on psychiatric and neurological disorders? A focus on CAPE and pinocembrin components. Medicinal Research Reviews 2020.
- Ağrı, İ.; Erdal Ağrı, A.; Özdemir, D.; Özgür, A. Chapter 31 - CAPE and Tympanosclerosis. In Polyphenols: Mechanisms of Action in Human Health and Disease (Second Edition), Watson, R.R., Preedy, V.R., Zibadi, S., Eds. Academic Press: 2018; https://doi.org/10.1016/B978-0-12-813006-3.00031-3pp. 421-430.
- Sforcin, J.M.; Bankova, V. Propolis: is there a potential for the development of new drugs? Journal of ethnopharmacology 2011, 133, 253-260.
- Toreti, V.C.; Sato, H.H.; Pastore, G.M.; Park, Y.K. Recent progress of propolis for its biological and chemical compositions and its botanical origin. Evidence-based complementary and alternative medicine 2013, 2013.
- Akyol, S.; Armutcu, F.; Yigitoglu, M. The medical usage of caffeic acid phenethyl ester (CAPE), an active compound of propolis, in neurological disorders and emergencies. Spatula DD 2011, 1, 37-42.
- Göçer, H.; Gülçin, İ. Caffeic acid phenethyl ester (CAPE): correlation of structure and antioxidant properties. International Journal of Food Sciences and Nutrition 2011, 62, 821-825.
- Tolba, M.F.; Azab, S.S.; Khalifa, A.E.; Abdel‐Rahman, S.Z.; Abdel‐Naim, A.B. Caffeic acid phenethyl ester, a promising component of propolis with a plethora of biological activities: A review on its anti‐inflammatory, neuroprotective, hepatoprotective, and cardioprotective effects. IUBMB life 2013, 65, 699-709.
- Murtaza, G.; Karim, S.; Akram, M.R.; Khan, S.A.; Azhar, S.; Mumtaz, A.; Bin Asad, M.H.H. Caffeic acid phenethyl ester and therapeutic potentials. BioMed Research International 2014, 2014.
- Eşrefoğlu, M.; Gül, M.; Ateş, B.; Yılmaz, İ. The ultrastructural and biochemical evidences of the beneficial effects of chronic caffeic acid phenethyl ester and melatonin administration on brain and cerebellum of aged rats. Fundamental & clinical pharmacology 2010, 24, 305-315.
- Amodio, R.; De Ruvo, C.; Di Matteo, V.; Poggi, A.; Di Santo, A.; Martelli, N.; Lorenzet, R.; Rotilio, D.; Cacchio, M.; Esposito, E. Caffeic acid phenethyl ester blocks apoptosis induced by low potassium in cerebellar granule cells. International journal of developmental neuroscience 2003, 21, 379-389.
- Ilhan, A.; Iraz, M.; Gurel, A.; Armutcu, F.; Akyol, O. Caffeic acid phenethyl ester exerts a neuroprotective effect on CNS against pentylenetetrazol-induced seizures in mice. Neurochemical Research 2004, 29, 2287-2292.
- Lin, M.-W.; Yang, S.-R.; Huang, M.-H.; Wu, S.-N. Stimulatory actions of caffeic acid phenethyl ester, a known inhibitor of NF-κB activation, on Ca2+-activated K+ current in pituitary GH3 cells. Journal of Biological Chemistry 2004, 279, 26885-26892.
- Yiş, U.; Topçu, Y.; Özbal, S.; Tuğyan, K.; Bayram, E.; Karakaya, P.; Yilmaz, O.; Kurul, S.H. Caffeic acid phenethyl ester prevents apoptotic cell death in the developing rat brain after pentylenetetrazole-induced status epilepticus. Epilepsy & Behavior 2013, 29, 275-280.
- Wei, X.; Ma, Z.; Fontanilla, C.; Zhao, L.; Xu, Z.; Taggliabraci, V.; Johnstone, B.; Dodel, R.; Farlow, M.; Du, Y. Caffeic acid phenethyl ester prevents cerebellar granule neurons (CGNs) against glutamate-induced neurotoxicity. Neuroscience 2008, 155, 1098-1105.
- dos Santos, N.A.G.; Martins, N.M.; de Barros Silva, R.; Ferreira, R.S.; Sisti, F.M.; dos Santos, A.C. Caffeic acid phenethyl ester (CAPE) protects PC12 cells from MPP+ toxicity by inducing the expression of neuron-typical proteins. Neurotoxicology 2014, 45, 131-138.
- Montpied, P.; de Bock, F.; Rondouin, G.; Niel, G.; Briant, L.; Courseau, A.-S.; Lerner-Natoli, M.; Bockaert, J. Caffeic acid phenethyl ester (CAPE) prevents inflammatory stress in organotypic hippocampal slice cultures. Molecular brain research 2003, 115, 111-120.
- Choi, K.; Choi, C. Differential regulation of c‐Jun N‐terminal kinase and NF‐κB pathway by caffeic acid phenethyl ester in astroglial and monocytic cells. Journal of neurochemistry 2008, 105, 557-564.
- Singh, J.; Khan, M.; Singh, I. Caffeic acid phenethyl ester induces adrenoleukodystrophy (Abcd2) gene in human X-ALD fibroblasts and inhibits the proinflammatory response in Abcd1/2 silenced mouse primary astrocytes. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids 2013, 1831, 747-758.
- Ilhan, A.; Akyol, O.; Gurel, A.; Armutcu, F.; Iraz, M.; Oztas, E. Protective effects of caffeic acid phenethyl ester against experimental allergic encephalomyelitis-induced oxidative stress in rats. Free radical biology and medicine 2004, 37, 386-394.
- Barber, S.C.; Higginbottom, A.; Mead, R.J.; Barber, S.; Shaw, P.J. An in vitro screening cascade to identify neuroprotective antioxidants in ALS. Free Radical Biology and Medicine 2009, 46, 1127-1138.
- Fontanilla, C.; Wei, X.; Zhao, L.; Johnstone, B.; Pascuzzi, R.; Farlow, M.; Du, Y. Caffeic acid phenethyl ester extends survival of a mouse model of amyotrophic lateral sclerosis. Neuroscience 2012, 205, 185-193.
- Tsai, C.-F.; Kuo, Y.-H.; Yeh, W.-L.; Wu, C.Y.-J.; Lin, H.-Y.; Lai, S.-W.; Liu, Y.-S.; Wu, L.-H.; Lu, J.-K.; Lu, D.-Y. Regulatory effects of caffeic acid phenethyl ester on neuroinflammation in microglial cells. International journal of molecular sciences 2015, 16, 5572-5589.
- Noelker, C.; Bacher, M.; Gocke, P.; Wei, X.; Klockgether, T.; Du, Y.; Dodel, R. The flavanoide caffeic acid phenethyl ester blocks 6-hydroxydopamine-induced neurotoxicity. Neuroscience letters 2005, 383, 39-43.
- Ma, Z.; Wei, X.; Fontanilla, C.; Noelker, C.; Dodel, R.; Hampel, H.; Du, Y. Caffeic acid phenethyl ester blocks free radical generation and 6-hydroxydopamine-induced neurotoxicity. Life sciences 2006, 79, 1307-1311.
- Silva, R.B.; Santos, N.; Martins, N.; Ferreira, D.; Barbosa Jr, F.; Souza, V.O.; Kinoshita, A.; Baffa, O.; Del-Bel, E.; Santos, A. Caffeic acid phenethyl ester protects against the dopaminergic neuronal loss induced by 6-hydroxydopamine in rats. Neuroscience 2013, 233, 86-94.
- Kurauchi, Y.; Hisatsune, A.; Isohama, Y.; Mishima, S.; Katsuki, H. Caffeic acid phenethyl ester protects nigral dopaminergic neurons via dual mechanisms involving haem oxygenase‐1 and brain‐derived neurotrophic factor. British journal of pharmacology 2012, 166, 1151-1168.
- Zaitone, S.A.; Ahmed, E.; Elsherbiny, N.M.; Mehanna, E.T.; El-Kherbetawy, M.K.; ElSayed, M.H.; Alshareef, D.M.; Moustafa, Y.M. Caffeic acid improves locomotor activity and lessens inflammatory burden in a mouse model of rotenone-induced nigral neurodegeneration: Relevance to Parkinson’s disease therapy. Pharmacological Reports 2019, 71, 32-41.
- Deveci, H.A.; Karapehlivan, M. Chlorpyrifos-induced parkinsonian model in mice: Behavior, histopathology and biochemistry. Pesticide biochemistry and physiology 2018, 144, 36-41.
- Fontanilla, C.; Ma, Z.; Wei, X.; Klotsche, J.; Zhao, L.; Wisniowski, P.; Dodel, R.; Farlow, M.; Oertel, W.; Du, Y. Caffeic acid phenethyl ester prevents 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine-induced neurodegeneration. Neuroscience 2011, 188, 135-141.
- Kumar, M.; Bansal, N. Caffeic acid phenethyl ester rescued streptozotocin-induced memory loss through PI3-kinase dependent pathway. Biomedicine & Pharmacotherapy 2018, 101, 162-173.
- Kumar, M.; Kaur, D.; Bansal, N. Caffeic acid phenethyl ester (CAPE) prevents development of STZ-ICV induced dementia in rats. Pharmacognosy magazine 2017, 13, S10.
- Morroni, F.; Sita, G.; Graziosi, A.; Turrini, E.; Fimognari, C.; Tarozzi, A.; Hrelia, P. Neuroprotective effect of caffeic acid phenethyl ester in a mouse model of Alzheimer’s disease involves Nrf2/HO-1 pathway. Aging and disease 2018, 9, 605.
- Ozyurt, B.; Ozyurt, H.; Akpolat, N.; Erdogan, H.; Sarsilmaz, M. Oxidative stress in prefrontal cortex of rat exposed to MK-801 and protective effects of CAPE. Progress in Neuro-Psychopharmacology and Biological Psychiatry 2007, 31, 832-838.
- Celik, S.; Erdogan, S. Caffeic acid phenethyl ester (CAPE) protects brain against oxidative stress and inflammation induced by diabetes in rats. Molecular and cellular biochemistry 2008, 312, 39-46.
- Korish, A.A.; Arafa, M.M. Propolis derivatives inhibit the systemic inflammatory response and protect hepatic and neuronal cells in acute septic shock. The Brazilian Journal of Infectious Diseases 2011, 15, 332-338.
- Fadillioglu, E.; Gursul, C.; Iraz, M. Effects of caffeic acid phenethyl ester on thioacetamide-induced hepatic encephalopathy in rats. Progress in Neuro-Psychopharmacology and Biological Psychiatry 2010, 34, 1440-1445.
- Jia, Y.; Jiang, S.; Chen, C.; Lu, G.; Xie, Y.; Sun, X.; Huang, L. Caffeic acid phenethyl ester attenuates nuclear factor‑κB‑mediated inflammatory responses in Müller cells and protects against retinal ganglion cell death. Molecular medicine reports 2019, 19, 4863-4871.
- Uzar, E.; Sahin, O.; Koyuncuoglu, H.R.; Uz, E.; Bas, O.; Kilbas, S.; Yilmaz, H.R.; Yurekli, V.A.; Kucuker, H.; Songur, A. The activity of adenosine deaminase and the level of nitric oxide in spinal cord of methotrexate administered rats: protective effect of caffeic acid phenethyl ester. Toxicology 2006, 218, 125-133.
- Uzar, E.; Koyuncuoglu, H.R.; Uz, E.; Yilmaz, H.R.; Kutluhan, S.; Kilbas, S.; Gultekin, F. The activities of antioxidant enzymes and the level of malondialdehyde in cerebellum of rats subjected to methotrexate: protective effect of caffeic acid phenethyl ester. Molecular and cellular biochemistry 2006, 291, 63-68.
- Uzar, E.; Koyuncuoğlu, H.R.; Yılmaz, H.R.; Uz, E.; Songur, A.; Şahin, Ö.; Yürekli, V.A.; Yılmaz, M.; Kılbaş, S.; Kutluhan, S. Ameliorating Role of Caffeic Acid Phenethyl Ester (CAPE) Against Methotrexate-Induced Oxidative Stress in the Sciatic Nerve, Spinal Cord and Brain Stem Tissues of Rats. Turkish Journal of Neurology/Turk Noroloji Dergisi 2010, 16.
- Ginis, Z.; Ozturk, G.; Albayrak, A.; Kurt, S.N.; Albayrak, M.; Fadillioglu, E. Protective effects of caffeic acid phenethyl ester on ifosfamide-induced central neurotoxicity in rats. Toxicology and Industrial Health 2016, 32, 337-343.
- Ferreira, R.S.; Dos Santos, N.A.G.; Martins, N.M.; Fernandes, L.S.; Dos Santos, A.C. Caffeic Acid Phenethyl Ester (CAPE) Protects PC12 Cells from Cisplatin-Induced Neurotoxicity by Activating the NGF-Signaling Pathway. Neurotoxicity research 2018, 34, 32-46, doi:10.1007/s12640-017-9849-z.
- Ferreira, R.S.; dos Santos, N.A.G.; Bernardes, C.P.; Sisti, F.M.; Amaral, L.; Fontana, A.C.; dos Santos, A.C. Caffeic acid phenethyl ester (CAPE) protects PC12 cells against cisplatin-induced neurotoxicity by activating the AMPK/SIRT1, MAPK/Erk, and PI3k/Akt signaling pathways. Neurotoxicity research 2019, 36, 175-192.
- Fadillioglu, E.; Erdogan, H.; Iraz, M.; Yagmurca, M. Effects of caffeic acid phenethyl ester against doxorubicin‐induced neuronal oxidant injury. Neuroscience Research Communications 2003, 33, 132-138.
- Eser, O.; Cosar, M.; Sahin, O.; Mollaoglu, H.; Sezer, M.; Yaman, M.; Songur, A. The neuroprotective effects of caffeic acid phenethyl ester (CAPE) in the hippocampal formation of cigarette smoke exposed rabbits. Pathology 2007, 39, 433-437.
- Başarslan, S.K.; Ösün, A.; Şenol, S.; Korkmaz, M.; Özkan, Ü.; Kaplan, I. Protective effects of intralipid and caffeic acid phenyl esther (CAPE) on neurotoxicity induced by ethanol in rats. 2017.
- Huang, Y.; Jin, M.; Pi, R.; Zhang, J.; Chen, M.; Ouyang, Y.; Liu, A.; Chao, X.; Liu, P.; Liu, J. Protective effects of caffeic acid and caffeic acid phenethyl ester against acrolein-induced neurotoxicity in HT22 mouse hippocampal cells. Neuroscience letters 2013, 535, 146-151.
- Mahmoud, A.M.; Abd El-Twab, S.M. Caffeic acid phenethyl ester protects the brain against hexavalent chromium toxicity by enhancing endogenous antioxidants and modulating the JAK/STAT signaling pathway. Biomedicine & Pharmacotherapy 2017, 91, 303-311.
- Hao, R.; Song, X.; Li, F.; Tan, X.; Sun-Waterhouse, D.; Li, D. Caffeic acid phenethyl ester reversed cadmium-induced cell death in hippocampus and cortex and subsequent cognitive disorders in mice: Involvements of AMPK/SIRT1 pathway and amyloid-tau-neuroinflammation axis. Food and Chemical Toxicology 2020, 144, 111636.
- Ozkan, U.; Osun, A.; Basarslan, K.; Senol, S.; Kaplan, I.; Alp, H. Effects of intralipid and caffeic acid phenethyl ester on neurotoxicity, oxidative stress, and acetylcholinesterase activity in acute chlorpyriphos intoxication. International Journal of Clinical and Experimental Medicine 2014, 7, 837.
- Uzar, E.; Varol, S.; Acar, A.; Firat, U.; Basarslan, S.; Evliyaoglu, O.; Yucel, Y.; Alp, H.; Gökalp, O. Assesment the role of oxidative stress and efficacy of caffeic acid phenethyl ester (CAPE) on neurotoxicity induced by isoniazid and ethambutol in a rat model. Eur Rev Med Pharmacol Sci 2014, 18, 2953-2959.
- Wang, L.Y.; Tang, Z.J.; Han, Y.Z. Neuroprotective effects of caffeic acid phenethyl ester against sevoflurane‑induced neuronal degeneration in the hippocampus of neonatal rats involve MAPK and PI3K/Akt signaling pathways. Molecular medicine reports 2016, 14, 3403-3412.
- Irmak, M.K.; Fadillioglu, E.; Sogut, S.; Erdogan, H.; Gulec, M.; Ozer, M.; Yagmurca, M.; Gozukara, M.E. Effects of caffeic acid phenethyl ester and alpha‐tocopherol on reperfusion injury in rat brain. Cell Biochemistry and Function: Cellular biochemistry and its modulation by active agents or disease 2003, 21, 283-289.
- Tsai, S.-K.; Lin, M.-J.; Liao, P.-H.; Yang, C.-Y.; Lin, S.-M.; Liu, S.-M.; Lin, R.-H.; Chih, C.-L.; Huang, S.-S. Caffeic acid phenethyl ester ameliorates cerebral infarction in rats subjected to focal cerebral ischemia. Life sciences 2006, 78, 2758-2762.
- Khan, M.; Elango, C.; Ansari, M.A.; Singh, I.; Singh, A.K. Caffeic acid phenethyl ester reduces neurovascular inflammation and protects rat brain following transient focal cerebral ischemia. Journal of neurochemistry 2007, 102, 365-377.
- Altuğ, M.E.; Serarslan, Y.; Bal, R.; Kontaş, T.; Ekici, F.; Melek, I.M.; Aslan, H.; Duman, T. Caffeic acid phenethyl ester protects rabbit brains against permanent focal ischemia by antioxidant action: a biochemical and planimetric study. Brain research 2008, 1201, 135-142.
- Serarslan, Y.; Bal, R.; Altuğ, M.E.; Kontaş, T.; Melek, I.M. CAFFEIC ACID PHENETHYL ESTER DECREASES THE LEVEL OF S-100B PROTEIN AFTER MIDDLE CEREBRAL AFTER OCCLUSION IN RABBITS. Pakistan journal of pharmaceutical sciences 2009, 22.
- Cengiz, N.; Colakoglu, N.; Kavakli, A.; Sahna, E.; Parlakpinar, H.; Acet, A. Effects of caffeic acid phenethyl ester on cerebral cortex: structural changes resulting from middle cerebral artery ischemia reperfusion. Clinical neuropathology 2007, 26, 80.
- Hwang, S.; Kim, C.D.; Lee, W.S. Caffeic acid phenethyl ester protects against photothrombotic cortical ischemic injury in mice. The Korean Journal of Physiology & Pharmacology 2018, 22, 101-110.
- Feng, Y.; Lu, Y.-W.; Xu, P.-H.; Long, Y.; Wu, W.-M.; Li, W.; Wang, R. Caffeic acid phenethyl ester and its related compounds limit the functional alterations of the isolated mouse brain and liver mitochondria submitted to in vitro anoxia–reoxygenation: relationship to their antioxidant activities. Biochimica et Biophysica Acta (BBA)-General Subjects 2008, 1780, 659-672.
- Wei, X.; Zhao, L.; Ma, Z.; Holtzman, D.M.; Yan, C.; Dodel, R.C.; Hampel, H.; Oertel, W.; Farlow, M.R.; Du, Y. Caffeic acid phenethyl ester prevents neonatal hypoxic–ischaemic brain injury. Brain 2004, 127, 2629-2635.
- Aladag, M.A.; Turkoz, Y.; Ozcan, C.; Sahna, E.; Parlakpınar, H.; Akpolat, N.; Cigremis, Y. Caffeic acid phenethyl ester (CAPE) attenuates cerebral vasospasm after experimental subarachnoidal haemorrhage by increasing brain nitric oxide levels. International journal of developmental neuroscience 2006, 24, 9-14.
- Palaz, M.N.; Akcay, E. The Impact of Propolis Factor Caffeic Acid Phenethyl-Ester on the Cerebral Vasospasm and Early Brain Damage in the Exparimentally Induced Subarachnoid Hemorrhage on Rats. World Neurosurgery 2020.
- Ilhan, A.; Koltuksuz, U.; Ozen, S.; Uz, E.; Ciralik, H.; Akyol, O. The effects of caffeic acid phenethyl ester (CAPE) on spinal cord ischemia/reperfusion injury in rabbits. European journal of cardio-thoracic surgery 1999, 16, 458-463.
- Ak, H.; Gulsen, I.; Karaaslan, T.; Alaca, İ.; Candan, A.; Koçak, H.; Atalay, T.; Çelikbilek, A.; Demir, İ.; Yılmaz, T. The effects of caffeic acid phenethyl ester on inflammatory cytokines after acute spinal cord injury. Ulus Travma Acil Cerrahi Derg 2015, 21, 96-101.
- Aydin, H.E.; Ozkara, E.; Ozbek, Z.; Vural, M.; Burukoglu, D.; Arslantas, A.; Atasoy, M.A. Histopathological evaluation of the effects of CAPE in experimental spinal cord injury. Turk Neurosurg 2016, 26, 437-444.
- Akgun, B.; Ozturk, S.; Artas, G.; Erol, F.S. Effects of intrathecal caffeic acid phenethyl ester (CAPE) on IL-6 and TNF-α levels and local inflammatory responses in spinal cord injuries. Turkish neurosurgery 2018, 28.
- Gocmez, C.; Celik, F.; Kamasak, K.; Kaplan, M.; Uzar, E.; Arıkanoglu, A.; Evliyaoglu, O. Effects of intrathecal caffeic acid phenethyl ester and methylprednisolone on oxidant/antioxidant status in traumatic spinal cord injuries. Journal of Neurological Surgery Part A: Central European Neurosurgery 2015, 76, 20-24.
- Kasai, M.; Fukumitsu, H.; Soumiya, H.; Furukawa, S. Caffeic acid phenethyl ester reduces spinal cord injury-evoked locomotor dysfunction. Biomedical Research 2011, 32, 1-7.
- Nasution, R.A.; Islam, A.A.; Hatta, M. Decreased neutrophil levels in mice with traumatic brain injury after cape administration. Annals of Medicine and Surgery 2020.
- Zhao, J.; Pati, S.; Redell, J.B.; Zhang, M.; Moore, A.N.; Dash, P.K. Caffeic acid phenethyl ester protects blood–brain barrier integrity and reduces contusion volume in rodent models of traumatic brain injury. Journal of neurotrauma 2012, 29, 1209-1218.
- Kerman, M.; Kanter, M.; Coşkun, K.K.; Erboga, M.; Gurel, A. Neuroprotective effects of caffeic acid phenethyl ester on experimental traumatic brain injury in rats. Journal of Molecular Histology 2012, 43, 49-57.
- Khan, M.; Shunmugavel, A.; Dhammu, T.S.; Khan, H.; Singh, I.; Singh, A.K. Combined treatment with GSNO and CAPE accelerates functional recovery via additive antioxidant activities in a mouse model of TBI. Journal of neuroscience research 2018, 96, 1900-1913.
- National Cancer Institute (Cancer Stat Facts: Brain and Other Nervous System Cancer). Availabe online: https://seer.cancer.gov/statfacts/html/brain.html (accessed on 3 November 2020).
- Choi, K.; Han, Y.-H.; Choi, C. N-Acetyl cysteine and caffeic acid phenethyl ester sensitize astrocytoma cells to Fas-mediated cell death in a redox-dependent manner. Cancer letters 2007, 257, 79-86.
- Park, M.H.; Kang, D.W.; Jung, Y.; Choi, K.-Y. Caffeic acid phenethyl ester downregulates phospholipase D1 via direct binding and inhibition of NFκB transactivation. Biochemical and biophysical research communications 2013, 442, 1-7.
- Morin, P.; St-Coeur, P.-D.; Doiron, J.A.; Cormier, M.; Poitras, J.J.; Surette, M.E.; Touaibia, M. Substituted caffeic and ferulic acid phenethyl esters: synthesis, leukotrienes biosynthesis inhibition, and cytotoxic activity. Molecules 2017, 22, 1124.
- Demestre, M.; Messerli, S.; Celli, N.; Shahhossini, M.; Kluwe, L.; Mautner, V.; Maruta, H. CAPE (caffeic acid phenethyl ester)‐based propolis extract (Bio 30) suppresses the growth of human neurofibromatosis (NF) tumor xenografts in mice. Phytotherapy Research: An International Journal Devoted to Pharmacological and Toxicological Evaluation of Natural Product Derivatives 2009, 23, 226-230.
- Wu, B.; Hao, Y.; Chen, Y.; Liu, Q.; Tian, C.; Zhang, Z.; Liu, J.; Wang, X. Studies on the structure-activity relationship of caffeate derivatives as neuroprotective agents. Journal of Chinese Pharmaceutical Sciences 2019, 28, 615-626, doi:10.5246/jcps.2019.09.059.
- Shi, H.; Xie, D.; Yang, R.; Cheng, Y. Synthesis of caffeic acid phenethyl ester derivatives, and their cytoprotective and neuritogenic activities in PC12 cells. Journal of agricultural and food chemistry 2014, 62, 5046-5053.
- Fu, W.; Wang, H.; Ren, X.; Yu, H.; Lei, Y.; Chen, Q. Neuroprotective effect of three caffeic acid derivatives via ameliorate oxidative stress and enhance PKA/CREB signaling pathway. Behavioural Brain Research 2017, 328, 81-86.
- Wan, T.; Wang, Z.; Luo, Y.; Zhang, Y.; He, W.; Mei, Y.; Xue, J.; Li, M.; Pan, H.; Li, W. FA-97, a New Synthetic Caffeic Acid Phenethyl Ester Derivative, Protects against Oxidative Stress-Mediated Neuronal Cell Apoptosis and Scopolamine-Induced Cognitive Impairment by Activating Nrf2/HO-1 Signaling. Oxidative medicine and cellular longevity 2019, 2019.
- Ning, X.; Guo, Y.; Ma, X.; Zhu, R.; Tian, C.; Zhang, Z.; Wang, X.; Ma, Z.; Liu, J. Design, synthesis and pharmacological evaluation of (E)-3, 4-dihydroxy styryl sulfonamides derivatives as multifunctional neuroprotective agents against oxidative and inflammatory injury. Bioorganic & medicinal chemistry 2013, 21, 5589-5597.
- Lu, D.-Y.; Huang, B.-R.; Yeh, W.-L.; Lin, H.-Y.; Huang, S.-S.; Liu, Y.-S.; Kuo, Y.-H. Anti-neuroinflammatory effect of a novel caffeamide derivative, KS370G, in microglial cells. Molecular neurobiology 2013, 48, 863-874.
- Silva, T.; Mohamed, T.; Shakeri, A.; Rao, P.P.; Martínez-González, L.; Pérez, D.I.; Martínez, A.; Valente, M.J.o.; Garrido, J.; Uriarte, E. Development of blood–brain barrier permeable nitrocatechol-based catechol O-methyltransferase inhibitors with reduced potential for hepatotoxicity. Journal of medicinal chemistry 2016, 59, 7584-7597.
- Silva, T.; Mohamed, T.; Shakeri, A.; Rao, P.P.; da Silva, P.S.; Remião, F.; Borges, F. Repurposing nitrocatechols: 5-Nitro-α-cyanocarboxamide derivatives of caffeic acid and caffeic acid phenethyl ester effectively inhibit aggregation of tau-derived hexapeptide AcPHF6. European journal of medicinal chemistry 2019, 167, 146-152.
This entry is adapted from the peer-reviewed paper 10.3390/biom11020176