Antibody-mediated disruption of the programmed cell death protein 1 (PD-1) pathway has brought much success to the fight against cancer. Nevertheless, a significant proportion of patients respond poorly to anti-PD-1 treatment. Cases of accelerated and more aggressive forms of cancer following therapy have also been reported. Termed hyper-progressive disease (HPD), this phenomenon often results in fatality, thus requires urgent attention. Among possible causes of HPD, regulatory T-cells (Tregs) are of suspect due to their high expression of PD-1, which modulates Treg activity. Tregs are a subset of CD4+ T-cells that play a non-redundant role in the prevention of autoimmunity and is functionally dependent on the X chromosome-linked transcription factor FoxP3. In cancer, CD4+FoxP3+ Tregs migrate to tumors to suppress anti-tumor immune responses, allowing cancer cells to persist.
- cancer
- Tregs
- immunosuppression
- PD-1
- hyper-progressive disease
1. Introduction
Cancer treatment has made great strides in recent years due to the advent of immune checkpoint blockade therapy. More research is embracing the concept of reinvigorating cancer-killing T-cells inactivated by co-inhibitory molecules. Programmed cell death-1 (PD-1) is, by far, the target of choice given that its expression is closely associated with T-cell dormancy and exhaustion. Indeed, enhanced tumor rejection from combining anti-PD-1 and conventional therapies has been reported [1][2]. Nevertheless, there remains a significant percentage of non-responders. Much work is still required to address the shortcomings and even unintended consequences of using anti-PD-1 therapy. One example of the latter is hyper-progressive disease (HPD).
HPD is a severe condition of cancer that is characterized by an acute acceleration in tumor growth following immunotherapy [3][4][5]. This was exemplified by a higher incidence of HPD among non-small cell lung cancer patients who received anti-PD-1 treatment (13.8%) compared to chemotherapy (5.1%) [6]. Although the overall rate of HPD occurrence seems to be fairly low at 10%, this varies widely with the type of cancer, ranging from an average of 9% in melanoma and others to 29% in head and neck squamous cell carcinoma. This partly stems from a lack of universal criteria for diagnosing HPD. Current methods differ on various factors such as tumor variable (diameter versus volume), rate of tumor growth (1.2 to 2 fold) and cut-off time to treatment failure [4][5][7]. There is, however, a clear consensus that HPD is an existential threat among anti-PD-1 treated cancer patients and that it often leads to a fatal outcome.
As more data become available, reasons behind this phenomenon would come to light and allow pre-emptive measures to be taken to mitigate HPD. Meanwhile, MDM2 and EGFR mutations have been flagged as potential risks rather than culprits, on the account that the oncogenic nature of these gene alterations predispose tumors to progress [8][9]. Hence, current efforts are dedicated to determine the exact cause of anti-PD-1-induced HPD. To date, two mechanisms have been proposed. The first involves the Fc region of anti-PD-1 antibody instigating M2-like differentiation of tumor-associated macrophages, thus cultivating immunosuppressive conditions in the tumor [10]. The second concerns regulatory T-cells (Tregs), the focus of this review.
Tregs are major perpetrators of cancer. Yet, there is considerable apprehension over the usage of Treg-targeted immunotherapy. This mainly arises from risks of adverse autoimmune reactions. For anti-PD-1 therapy, perhaps a calibrated approach would be needed as it is becoming evident that Tregs may not only reduce its efficacy but also bring about HPD. This is supported by evidence of higher Treg levels in the peripheral blood of non-responders and a recent claim that higher frequency of PD-1+ effector T-cells relative to PD-1+ Tregs in the tumor predicts positive response to therapy [11][12]. Moreover, increased immunosuppression by Tregs that lack PD-1 signaling has been shown to accelerate tumor development in mice modeled on HPD [13].
2. From Friend to Foe—Regulatory T-Cell (Treg) Induction of Cancer Immune Tolerance
2.1. Tregs in Autoimmunity and Cancer
The majority of CD4+ Tregs develop in the thymus and constitute about 10% of circulating CD4+ T-cells. Tregs play a non-redundant role in immune tolerance and have a master transcription factor, Foxp3, which largely defines the phenotypic and functional characteristics of Tregs. Mutation of FoxP3 gene results in immunodysregulation polyendocrinopathy enteropathy X-linked syndrome in humans and scurfy in mice [14][15][16]. While Tregs are indispensable to protection against autoimmunity, they are undesirable to cancer immunity. A high frequency of Tregs in the tumor usually spells poor prognosis [17]. There are a few exceptions marked by favorable outcomes possibly as a result of Tregs responding but failing to contain strong anti-tumor responses [18]. A key notion is that the more immunogenic tumors are, the more they may be subjected to Treg immunosuppression. This is corroborated by numerous animal studies showing greatly reduced tumors after depleting Tregs or rendering Tregs defective in function [19][20][21]. It is also worth mentioning that selective elimination of intra-tumoral Tregs is sufficient to bolster cytotoxic killing of tumor cells without perturbing the systemic immune system. Using a technique called photodynamic therapy, this was demonstrated by aiming a laser beam at tumors to deplete only tumor-resident Tregs that were pre-bound with a photosensitizer-conjugated antibody against CD25, a dominant Treg surface marker [22]. A similar result from deleting glucocorticoid-induced tumor necrosis factor receptor (GITR)-expressing Tregs would have added more credence, on the grounds that anti-GITR treatment strongly reversed the growth of advanced tumors [23].
2.2. Recruitment of Tregs into Tumor
Several chemokine receptors and their partner chemokines have been implicated in the recruitment of Tregs to tumors. CCR4 emerged as a prime candidate after it was found to be expressed in Tregs of human ovarian cancer containing substantial amounts of its ligand, CCL22 [24]. With more precise analyses of the Treg compartment, CCR4 was later found to be present almost exclusively on activated, but not naïve, Tregs [25]. It was then no coincidence that anti-CCR4 treatment in patients with adult T-cell leukemia-lymphoma evoked strong tumor antigen-specific CD8+ T-cell response. This was in support of CCR4 blockade reducing tumor-infiltrating Tregs and enhancing anti-tumor immunity especially when combined with tumor vaccines [26][27].
In human primary breast cancer, Tregs appear to have a peculiar trafficking pattern as they are recruited by CCL22 to only the border of tumor not within [28][29]. The retention of Tregs at the boundary could be attributed to downregulation or internalization of CCR4 upon binding to CCL22, resulting in cessation of chemotaxis. Tregs in this region are highly activated and proliferative [29]. They may tolerize effector T-cells before their entry into tumor. It would be of interest to assess possible effects of anti-PD-1 treatment on the tumor sentry duties of these Tregs.
In pancreatic and squamous cell carcinoma, the CCL5:CCR5 axis serves as the main highway for Tregs to infiltrate into tumors [30][31]. Reducing CCL5 production by tumor cells or blocking CCR5 on Tregs significantly lessens the presence of Tregs in tumors. More importantly, CCR5 is preferentially expressed on CD4+Foxp3+ Tregs over CD4+Foxp3− T-cells in healthy individuals and more so in cancer patients [30]. This pattern of expression can be retrieved easily from cells in the peripheral blood and is, therefore, a viable and convenient biomarker.
Besides CCR4 and CCR5, other notable chemokine receptors include CCR8, CXCR3 and CXCR6. These were recently identified as signature genes of tumor-infiltrating Tregs through large-scale mining of Treg transcriptomes from various tumor models and tissues (e.g., colon and spleen) cross-checked between species (humans and mice) and with datasets from the human genome atlas [32]. Interestingly, in vivo CRISPR-Cas9 screen revealed that Tregs deficient in CXCR3, but not CCR8 and CXCR6, were reduced in tumors [32]. This suggests that CCR8 and CXCR6 do not participate in steering Tregs to tumors but may be upregulated by tumor-specific conditions. CCR8, in particular, has been identified as a promising target for specific depletion of intra-tumoral Tregs. Mice administered with anti-CCR8 antibody were shown to have diminished tumors that contained fewer Tregs and more inflammatory cell infiltrates [33]. Two questions that ought to be addressed henceforth are the main mechanism-of-action which CCR8+ Tregs employ to create an immunosuppressive environment and the location in tumors where they reside to do so. Additionally, determining the relative expression of PD-1 in CCR8+ Tregs and their response to PD-1 blockade would be insightful.
2.3. Metabolic Reprogramming of Tumor-Infiltrating Tregs
The tumor microenvironment (TME) is hypoxic and low in glucose. This presents a challenge to tumor-infiltrating T-cells which depend on glycolysis as their primary metabolic machinery. Tregs, however, have an intrinsic advantage with FoxP3 downregulating c-Myc to enable coupling to oxidative phosphorylation (OXPHOS) [34]. As opposed to other T-cells, Tregs display heightened OXPHOS underscored by higher mitochondrial activity and reactive oxygen species [35][36]. A key mediator of OXPHOS in Tregs is liver kinase B1 (LKB1). Tregs deficient in LKB1 are metabolically and functionally impaired owing to dysfunctional mitochondria and dysregulated β-catenin signaling that imposes high PD-1 expression [37]. Blocking PD-1 restored Treg suppression, indicating differential metabolic requirements by Tregs as they transit from one functional state to another.
The versatility of Tregs to adapt to the TME is also underpinned by increased expression of the mitochondrial enzyme, carnitine palmitoyltransferase 1a, which orchestrates the transport of acyl-CoA into mitochondria for fatty acid oxidation (FAO) [38][39]. Hence, Tregs can turn to lipids as a secondary source of energy. Several studies are supportive of fatty acid synthesis and FAO providing a metabolic advantage to Tregs over other T-cells within the tumor [40][41]. In patients with gastric cancer, resistance to PD-1 blockade therapy was traced to a mutation in tumor cells that elevated fatty acid production [42]. This favored Treg survival and prevalence, and was deduced to undermine the effectiveness of anti-PD-1 therapy.
3. Tumor-Associated Immunosuppressive Mechanisms of Tregs
Tregs suppress conventional CD4+ T-cells (Tconv) by multiple cell-contact and bystander mechanisms (Figure 1). They essentially ‘bubble wrap’ Tconv cells under several layers of regulatory control. This is an efficient and sustainable strategy for a minor to contain a major.
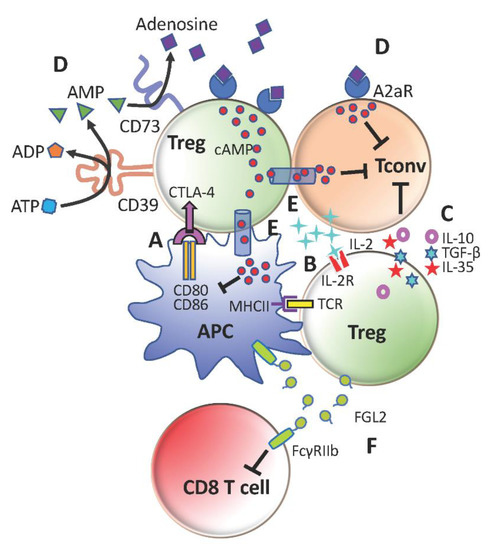
Figure 1. Immunosuppressive mechanisms of regulatory T-cells (Tregs). (A) Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)-mediated downregulation of CD80 and CD86 on antigen-presenting cells (APCs) to limit conventional CD4+ T-cells (Tconv) activation. (B) Compete with Tconv cells for IL-2 to restrict Tconv survival and proliferation. (C) Secretion of anti-inflammatory cytokines TGFb, IL-10 and IL-35 to promote Tconv exhaustion and repress Tconv effector functions (D) Generation of adenosine with CD39 and CD73 to suppress Tconv cells and further stimulate Tregs through A2aR (E) Transfer of cAMP to inhibit activation of Tconv cells and APCs (F) Production of FGL2 to inhibit CD8+ T-cells and APCs through FcγRIIb.
4. Tregs ‘Tipped’ by Anti-Programmed Cell Death Protein 1 (PD-1) Turn the Tables on Anti-Tumor T-Cells
4.1. PD-1 Inhibitory Signaling
When T-cells become activated, NFATc1 binds to the promoter of PD-1 to initiate transcription [43]. This is augmented on re-stimulation and is counterbalanced by the association of special AT-rich sequence binding protein 1 (Satb1) and nucleosome remodeling deacetylase (NURD) in the enhancer regions of PD-1 [44]. T-cells deficient in Satb1 promptly gain PD-1 and assume dormancy. In tumors, de-repression of PD-1 may occur with TGFβ-induced Smad proteins displacing the Satb1:NURD complex [44]. This could explain the especially high expression of PD-1 in tumor-infiltrating T-cells.
The cytoplasmic domain of PD-1 comprises two tyrosine-based residues, ITIM and immunoreceptor tyrosine-based switch motif (ITSM). Phosphorylation of ITSM recruits SH2-domain containing tyrosine phosphatase 2 (SHP-2) to dephosphorylate adaptor molecules of the TCR complex, such as CD3, zeta-chain-associated protein of 70kDa (ZAP70) and phosphatidylinositol-3-kinase (PI3K) [45]. This process may also relieve Csk from inhibition by SHP-2, allowing Csk to preclude lymphocyte-specific protein tyrosine kinase (Lck) from activating CD3 and ZAP70 [46].
4.2. Shortcomings of Anti-PD-1 Cancer Immunotherapy
Among the cancer types treated by PD-1 blockade therapy, Hodgkin’s disease has the highest objective response rate at 87%, while head and neck, gastroesophageal and bladder cancer share the lowest at 15% [47]. The tumor mutational burden ranks high on the list of factors that influence treatment efficacy [48][49]. It can be categorized by mutations that prevail from early tumor cells, termed clonal, or mutations that arise during tumor development, termed ‘subclonal’ [49]. In general, anti-PD-1 therapy is more effective against tumors that contain higher clonal than subclonal mutations. These tumors are likely to have a substantial pool of PD-1+ effector T-cells ready to be unleashed against clonal antigens that are ubiquitously expressed on tumor cells. On the other hand, a far from absolute response is expected of tumors with subclonal antigens. Tumor cells that escape from anti-PD-1 immunotherapy may thrive better after the destruction of their more vulnerable neighbors, particularly in a nutrient scarce tumor. This could precipitate tumor progression. Under such a circumstance, unless sufficient effector T-cells are generated against subclonal antigens, anti-PD-1 therapy would be less effective. Tregs could present a major hurdle too.
4.3. PD-1 Signaling Inhibits Treg Activity and Blockade of PD-1 Enhances Treg-Mediated Immunosuppression
In patients with glioblastoma multiforme, Tregs with high PD-1 expression are less suppressive and have a transcriptomic profile that bears an exhausted signature [50]. It was, likewise, the case for Tregs in patients infected with hepatitis C [51]. Blocking PD-1 in Tregs isolated from livers of these patients gave rise to increased Treg proliferation and suppression.
Consistent with PD-1 restricting Treg activity, CXCR5+ follicular Tregs from PD-1 deficient (PD-1KO) mice were found to be more immunosuppressive in vitro [52]. Correspondingly, adoptive transfer of PD-1KO Tregs into mice susceptible to autoimmune pancreatitis led to better suppression of pathogenic Tconv and CD8+ T-cells and better protection of the pancreas compared to the transfer of wild-type Tregs [53]. Reduced type 1 diabetes in non-obese diabetic (NOD) mice with PD-1 deficiency limited to only Tregs provided the strongest affirmation for a repressive role of PD-1 in Treg immunosuppression [54]. Tregs devoid of PD-1 were also shown to have reduced PI3K-AKT signaling that is typical of activated Tregs.
As with the intent of releasing the ‘brake’ on anti-tumor T-cells, PD-1 blockade therapy may still have to circumvent the pro-tumoral Treg barrier. Blockade of PD-1 could have a pronounced impact on Tregs as they constantly engage and receive TCR stimulation from APCs. In a Phase I trial of nivolumab administered to stage III/IV melanoma, an increase in Treg frequency was observed in leukapheresis specimens of non-responders [11]. In addition, animal tumor models resistant to anti-PD-1 became less so with concurrent depletion of Tregs using the anti-CD25 antibody [55].
More than offsetting tumor-killing effects, the expansion of Tregs that accompanies PD-1 blockade could instigate tumor progression akin to HPD (Figure 2). As previously reported by us, increased Treg proliferation was apparent in advanced gastric cancer patients diagnosed with HPD. We further verified in murine experiments that PD-1 deficiency and blockade in Tregs boosted their immunosuppressive action against even PD-1KO effector T-cells to advance tumor development [13]. Nonetheless, Tregs are unlikely to be the sole cause of HPD. Certain underlying preconditions may advance tumors to the verge of explosive growth before PD-1 blockade escalates the process through Treg activation. These could be present during the ‘equilibrium’ phase, defined as the stage where tumor eradication by effector T-cells is matched by tumor evasion [56]. At this point of inflection, any decrease in effector T-cell activity could benefit tumor cell survival and division. For instance, the metabolic rate of tumor cells may be spurred as more glucose becomes available. This is a reasonable speculation in view of the strict reliance on aerobic glycolysis by tumor cells and effector T-cells. All in all, anti-PD-1 may have less activated PD-1+ effector T-cells to target from increased Treg suppression.
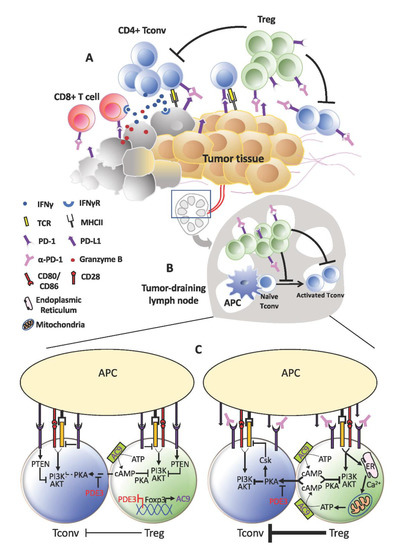
Figure 2. Treg expansion behind hyper-progressive disease (HPD). (A) Within the tumor, the anti-tumor effects of programmed cell death protein 1 (PD-1) blockade on effector T-cells may be short-lived due to the activation and expansion of Tregs. This could be even more pronounced in tumors that contain higher proportions of PD-1+ Tregs relative to T-effector cells. Consequently, tumor cells that escape continue to thrive and proliferate. (B) A similar occurrence may take place in the lymphoid organs where a more active Treg population resists fresh generation of anti-tumor T-effector cells. As anti-tumor immunity wanes, tumor progresses unhindered and may even accelerate in the form of HPD. (C) At the cellular level, increased TCR signaling in Tregs devoid of PD-1 signaling may drive increased calcium influx and adenosine triphosphate (ATP) production, which in turn elevates cAMP generation and transfer into Tconv cells. Consequently, TCR stimulation in Tconv cells could still be subdued by the PKA:Csk despite absence of PD-1 inhibitory signals.
5. Tracing the Path to Hyper-Progressive Disease (HPD) after Treg Expansion by PD-1 Blockade
While blocking the inhibitory signals of PD-1 increases Treg activation, Tregs may still require IL-2 for robust proliferation. A study by Asano and co. showed that Tregs, but not non-Treg T-cells, expanded after IL-2 was administered at a low dose to mice [57]. The number of Tregs rose sharply after treatment with anti-PD-1. This was followed by a quick return to baseline level due to Treg apoptosis. A similar trend was observed in PD-1KO mice.
In the context of anti-PD-1 cancer immunotherapy, Treg expansion may be sustained by transient release of IL-2 from effector T-cells in the initial phase. In time, IL-2 secretion could subside as effector T-cells become increasingly suppressed by Tregs. Discontinuation of IL-2 production in effector T-cells may also be brought forth by increased expression of other co-inhibitory molecules, such as Tim-3 (T-cell immunoglobulin and mucin domain-3) and Lag-3 (lymphocyte-activation gene 3) [58][59]. This could expedite exhaustion in effector T-cells and prevent them from re-committing to the killing of tumor cells.
Although the apoptotic rate of Tregs during anti-PD-1 treatment is not known, increased Treg apoptosis could potentiate tumor progression. This is based on the strong immunosuppressive effect of dying Tregs through adenosine production as mentioned above [60]. However, it is unlikely for extra adenosine to cause HPD simply by acting against effector T-cells that are already largely suppressed. Rather, adenosine has several pro-tumoral properties that could steepen the growth trajectory of tumor cells. Among them are improving tumor cell survivability through the regulation of anti- (Bcl2) and pro- (p53 and Bax) apoptotic genes and fostering proliferation through the induction of relevant signaling pathways (e.g., Akt and Erk1/2) as well as cell-cycle factors (e.g. cyclins A, B, D and E) [61]. Beyond these, adenosine may raise the chance of tumor malignancy by weakening the adhesion and stability of primary tumors and enhancing angiogenesis that provides conduits for tumor cells to emigrate and invade other tissues and organs [61][62][63]. Nevertheless, there are occasions where adenosine is linked to tumor growth arrest [61]. Variations in response to adenosine may stem from the subtypes (A1R, A2aR, A2bR, A3R) and levels of adenosine receptors expressed in tumor cells.
It is imperative not to overlook the possibility of Treg expansion causing HPD. It is equally imperative not to assume any degree of Treg expansion leads to HPD. Hypothetically, minor Treg expansion may only give rise to non-response to therapy or moderate tumor progression. Major Treg expansion, which may be contingent to IL-2 availability, and a subsequent ‘tsunami’ of Treg apoptosis plus adenosine production is more likely to culminate in HPD. Further work is needed to uncover the pathogenesis of Treg-mediated HPD. This is a difficult undertaking given the extremely short time frame of HPD.
At present, tumors assessed in accordance with RECIST1.1 with growth rate of more than two-fold at first examination compared to pre-therapy would qualify as HPD. Stricter criteria have been proposed to classify and distinguish HPD from other forms of cancer progression [64]. Some recommendations are the inclusion of time to treatment failure (TTF), measurement of the longest tumor dimension relative to time to better represent growth kinetics, additional radiological scans and detection of tumor metastasis [8][10][65][66]. Perhaps, these stringent guidelines may expose differences between HPD cases that are dependent and independent of Treg expansion. One may expect HPD driven by Treg expansion to have shorter TTF and prone to metastasis. Additionally, the growing list of HPD biomarkers can be stratified by the presence of Tregs in tumors [67] This may assist in identifying correlations between Tregs and certain tumor-associated features that have a higher propensity for HPD development. Dissecting Treg populations into viable and apoptotic Tregs and analyzing the variety of adenosine receptors in tumor biopsies would be desirable in this respect.
This entry is adapted from the peer-reviewed paper 10.3390/cancers13010048
References
- Dovedi, S.J.; Adlard, A.L.; Lipowska-Bhalla, G.; McKenna, C.; Jones, S.; Cheadle, E.J.; Stratford, I.J.; Poon, E.; Morrow, M.; Stewart, R.; et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. 2014, 74, 5458–5468.
- Yan, Y.; Kumar, A.B.; Finnes, H.; Markovic, S.N.; Park, S.; Dronca, R.S.; Dong, H. Combining immune checkpoint inhibitors with conventional cancer therapy. Front. Immunol. 2018, 9, 1739.
- Champiat, S.; Dercle, L.; Ammari, S.; Massard, C.; Hollebecque, A.; Postel-Vinay, S.; Chaput, N.; Eggermont, A.; Marabelle, A.; Soria, J.C.; et al. Hyperprogressive disease is a new pattern of progression in cancer patients treated by Anti-PD-1/PD-L1. Clin. Cancer Res. 2017, 23, 1920–1928.
- Champiat, S.; Ferrara, R.; Massard, C.; Besse, B.; Marabelle, A.; Soria, J.C.; Ferté, C. Hyperprogressive disease: Recognizing a novel pattern to improve patient management. Nat. Rev. Clin. Oncol. 2018, 15, 748–762.
- Borcoman, E.; Kanjanapan, Y.; Champiat, S.; Kato, S.; Servois, V.; Kurzrock, R.; Goel, S.; Bedard, P.; Le Tourneau, C. Novel patterns of response under immunotherapy. Ann. Oncol. 2019, 30, 385–396.
- Ferrara, R.; Mezquita, L.; Texier, M.; Lahmar, J.; Audigier-Valette, C.; Tessonnier, L.; Mazieres, J.; Zalcman, G.; Brosseau, S.; Le Moulec, S.; et al. Hyperprogressive disease in patients with advanced non-small cell lung cancer treated with PD-1/PD-L1 Inhibitors or with single-agent chemotherapy. JAMA Oncol. 2018, 4, 1543–1552.
- Popat, V.; Gerber, D.E. Hyperprogressive disease: A distinct effect of immunotherapy? J. Thorac. Dis. 2019, 11, S262–S265.
- Kato, S.; Goodman, A.; Walavalkar, V.; Barkauskas, D.A.; Sharabi, A.; Kurzrock, R. Hyperprogressors after immunotherapy: Analysis of genomic alterations associated with accelerated growth rate. Clin. Cancer Res. 2017, 23, 4242–4250.
- Adashek, J.J.; Subbiah, I.M.; Matos, I.; Garralda, E.; Menta, A.K.; Ganeshan, D.M.; Subbiah, V. Hyperprogression and immunotherapy: Fact, fiction, or alternative fact? Trends Cancer 2020, 6, 181–191.
- Lo Russo, G.; Moro, M.; Sommariva, M.; Cancila, V.; Boeri, M.; Centonze, G.; Ferro, S.; Ganzinelli, M.; Gasparini, P.; Huber, V.; et al. Antibody-Fc/FcR interaction on macrophages as a mechanism for hyperprogressive disease in non-small cell lung cancer subsequent to PD-1/PD-L1 blockade. Clin. Cancer Res. 2019, 25, 989–999.
- Weber, J.S.; Kudchadkar, R.R.; Yu, B.; Gallenstein, D.; Horak, C.E.; Inzunza, H.D.; Zhao, X.; Martinez, A.J.; Wang, W.; Gibney, G.; et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or-naive melanoma. J. Clin. Oncol. 2013, 31, 4311–4318.
- Kumagai, S.; Togashi, Y.; Kamada, T.; Sugiyama, E.; Nishinakamura, H.; Takeuchi, Y.; Vitaly, K.; Itahashi, K.; Maeda, Y.; Matsui, S.; et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat. Immunol. 2020, 21, 1346–1358.
- Kamada, T.; Togashi, Y.; Tay, C.; Ha, D.; Sasaki, A.; Nakamura, Y.; Sato, E.; Fukuoka, S.; Tada, Y.; Tanaka, A.; et al. PD-1+ regulatory T-cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 9999–10008.
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061.
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336.
- Khattri, R.; Cox, T.; Yasayko, S.A.; Ramsdell, F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 2003, 4, 337–342.
- Shang, B.; Liu, Y.; Jiang, S.J. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 15179.
- Echarti, A.; Hecht, M.; Büttner-Herold, M.; Haderlein, M.; Hartmann, A.; Fietkau, R.; Distel, L. CD8+ and regulatory T cells differentiate tumor immune phenotypes and predict survival in locally advanced head and neck cancer. Cancers 2019, 11, 1398.
- Onizuka, S.; Tawara, I.; Shimizu, J.; Sakaguchi, S.; Fujita, T.; Nakayama, E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res. 1999, 59, 3128–3133.
- Li, X.; Kostareli, E.; Suffner, J.; Garbi, N.; Hämmerling, G.J. Efficient Treg depletion induces T-cell infiltration and rejection of large tumors. Eur. J. Immunol. 2010, 40, 3325–3335.
- Klages, K.; Mayer, C.T.; Lahl, K.; Loddenkemper, C.; Teng, M.W.; Ngiow, S.F.; Smyth, M.J.; Hamann, A.; Huehn, J.; Sparwasser, T. Selective depletion of Foxp3+ regulatory T cells improves effective therapeutic vaccination against established melanoma. Cancer Res. 2010, 70, 7788–7799.
- Oh, D.S.; Kim, H.; Oh, J.E.; Jung, H.E.; Lee, Y.S.; Park, J.H.; Lee, H.K. Intratumoral depletion of regulatory T cells using CD25-targeted photodynamic therapy in a mouse melanoma model induces antitumoral immune responses. Oncotarget 2017, 8, 47440–47453.
- Ko, K.; Yamazaki, S.; Nakamura, K.; Nishioka, T.; Hirota, K.; Yamaguchi, T.; Shimizu, J.; Nomura, T.; Chiba, T.; Sakaguchi, S. Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J. Exp. Med. 2005, 202, 885–891.
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949.
- Sugiyama, D.; Nishikawa, H.; Maeda, Y.; Nishioka, M.; Tanemura, A.; Katayama, I.; Ezoe, S.; Kanakura, Y.; Sato, E.; Fukumori, Y.; et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc. Natl. Acad. Sci. USA 2013, 110, 17945–17950.
- Ishida, T.; Ishii, T.; Inagaki, A.; Yano, H.; Komatsu, H.; Iida, S.; Inagaki, H.; Ueda, R. Specific recruitment of CC chemokine receptor 4-positive regulatory T cells in Hodgkin lymphoma fosters immune privilege. Cancer Res. 2006, 66, 5716–5722.
- Pere, H.; Montier, Y.; Bayry, J.; Quintin-Colonna, F.; Merillon, N.; Dransart, E.; Badoual, C.; Gey, A.; Ravel, P.; Marcheteau, E.; et al. A CCR4 antagonist combined with vaccines induces antigen-specific CD8+ T cells and tumor immunity against self antigens. Blood 2011, 118, 4853–4862.
- Ménétrier-Caux, C.; Gobert, M.; Caux, C. Differences in tumor regulatory T-cell localization and activation status impact patient outcome. Cancer Res. 2009, 69, 7895–7898.
- Gobert, M.; Treilleux, I.; Bendriss-Vermare, N.; Bachelot, T.; Goddard-Leon, S.; Arfi, V.; Biota, C.; Doffin, A.C.; Durand, I.; Olive, D.; et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res. 2009, 69, 2000–2009.
- Tan, M.C.; Goedegebuure, P.S.; Belt, B.A.; Flaherty, B.; Sankpal, N.; Gillanders, W.E.; Eberlein, T.J.; Hsieh, C.S.; Linehan, D.C. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J. Immunol. 2009, 182, 1746–1755.
- de Oliveira, C.E.; Gasparoto, T.H.; Pinheiro, C.R.; Amôr, N.G.; Nogueira, M.R.S.; Kaneno, R.; Garlet, G.P.; Lara, V.S.; Silva, J.S.; Cavassani, K.A.; et al. CCR5-Dependent homing of T regulatory cells to the tumor microenvironment contributes to skin squamous cell carcinoma development. Mol. Cancer Ther. 2017, 16, 2871–2880.
- Magnuson, A.M.; Kiner, E.; Ergun, A.; Park, J.S.; Asinovski, N.; Ortiz-Lopez, A.; Kilcoyne, A.; Paoluzzi-Tomada, E.; Weissleder, R.; Mathis, D.; et al. Identification and validation of a tumor-infiltrating Treg transcriptional signature conserved across species and tumor types. Proc. Natl. Acad. Sci. USA 2018, 115, E10672–E10681.
- Villarreal, D.O.; L’Huillier, A.; Armington, S.; Mottershead, C.; Filippova, E.V.; Coder, B.D.; Petit, R.G.; Princiotta, M.F. Targeting CCR8 induces protective antitumor immunity and enhances vaccine-induced responses in colon cancer. Cancer Res. 2018, 78, 5340–5348.
- Angelin, A.; Gil-de-Gómez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J.; Kopinski, P.K.; Wang, L.; et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. 2017, 25, 1282–1293.e7.
- Beier, U.H.; Angelin, A.; Akimova, T.; Wang, L.; Liu, Y.; Xiao, H.; Koike, M.A.; Hancock, S.A.; Bhatti, T.R.; Han, R.; et al. Essential role of mitochondrial energy metabolism in Foxp3⁺ T-regulatory cell function and allograft survival. FASEB J. 2015, 29, 2315–2326.
- Pacella, I.; Procaccini, C.; Focaccetti, C.; Miacci, S.; Timperi, E.; Faicchia, D.; Severa, M.; Rizzo, F.; Coccia, E.M.; Bonacina, F.; et al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc. Natl. Acad. Sci. USA 2018, 115, E6546–E6555.
- Yang, K.; Blanco, D.B.; Neale, G.; Vogel, P.; Avila, J.; Clish, C.B.; Wu, C.; Shrestha, S.; Rankin, S.; Long, L.; et al. Homeostatic control of metabolic and functional fitness of Treg cells by LKB1 signaling. Nature 2017, 548, 602–606.
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303.
- Procaccini, C.; Carbone, F.; Di Silvestre, D.; Brambilla, F.; De Rosa, V.; Galgani, M.; Faicchia, D.; Marone, G.; Tramontano, D.; Corona, M.; et al. The proteomic landscape of human ex vivo regulatory and conventional T cells reveals specific metabolic requirements. Immunity 2016, 44, 712.
- Miska, J.; Lee-Chang, C.; Rashidi, A.; Muroski, M.E.; Chang, A.L.; Lopez-Rosas, A.; Zhang, P.; Panek, W.K.; Cordero, A.; Han, Y.; et al. HIF-1α is a metabolic switch between glycolytic-driven migration and oxidative phosphorylation-driven immunosuppression of Tregs in glioblastoma. Cell Rep. 2019, 27, 226–237.e4.
- Wang, H.; Franco, F.; Tsui, Y.C.; Xie, X.; Trefny, M.P.; Zappasodi, R.; Mohmood, S.R.; Fernández-García, J.; Tsai, C.H.; Schulze, I.; et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol. 2020, 21, 298–308.
- Kumagai, S.; Togashi, Y.; Sakai, C.; Kawazoe, A.; Kawazu, M.; Ueno, T.; Sato, E.; Kuwata, T.; Kinoshita, T.; Yamamoto, M.; et al. An oncogenic alteration creates a microenvironment that promotes tumor progression by conferring a metabolic advantage to regulatory T cells. Immunity 2020, 53, 187–203.e8.
- Oestreich, K.J.; Yoon, H.; Ahmed, R.; Boss, J.M. NFATc1 regulates PD-1 expression upon T cell activation. J. Immunol. 2008, 181, 4832–4839.
- Stephen, T.L.; Payne, K.K.; Chaurio, R.A.; Allegrezza, M.J.; Zhu, H.; Perez-Sanz, J.; Perales-Puchalt, A.; Nguyen, J.M.; Vara-Ailor, A.E.; Eruslanov, E.B.; et al. SATB1 expression governs epigenetic repression of PD-1 in tumor-reactive T cells. Immunity 2017, 46, 51–64.
- Patsoukis, N.; Wang, Q.; Strauss, L.; Boussiotis, V.A. Revisiting the PD-1 pathway. Sci. Adv. 2020, 6, eabd2712.
- Sheppard, K.A.; Fitz, L.J.; Lee, J.M.; Benander, C.; George, J.A.; Wooters, J.; Qiu, Y.; Jussif, J.M.; Carter, L.L.; Wood, C.R.; et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004, 574, 37–41.
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355.
- Kim, J.Y.; Kronbichler, A.; Eisenhut, M.; Hong, S.H.; van der Vliet, H.J.; Kang, J.; Shin, J.I.; Gamerith, G. Tumor mutational burden and efficacy of immune checkpoint inhibitors: A systematic review and meta-analysis. Cancers 2019, 11, 1798.
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469.
- Lowther, D.E.; Goods, B.A.; Lucca, L.E.; Lerner, B.A.; Raddassi, K.; van Dijk, D.; Hernandez, A.L.; Duan, X.; Gunel, M.; Coric, V.; et al. PD-1 marks dysfunctional regulatory T cells in malignant gliomas. JCI Insight 2016, 1, e85935.
- Franceschini, D.; Paroli, M.; Francavilla, V.; Videtta, M.; Morrone, S.; Labbadia, G.; Cerino, A.; Mondelli, M.U.; Barnaba, V. PD-L1 negatively regulates CD4+CD25+Foxp3+ Tregs by limiting STAT-5 phosphorylation in patients chronically infected with HCV. J. Clin. Investig. 2009, 119, 551–564.
- Sage, P.T.; Francisco, L.M.; Carman, C.V.; Sharpe, A.H. The receptor PD-1 controls follicular regulatory T cells in the lymph nodes and blood. Nat. Immunol. 2013, 14, 152–161.
- Zhang, B.; Chikuma, S.; Hori, S.; Fagarasan, S.; Honjo, T. Nonoverlapping roles of PD-1 and FoxP3 in maintaining immune tolerance in a novel autoimmune pancreatitis mouse model. Proc. Natl. Acad. Sci. USA 2016, 113, 8490–8495.
- Tan, C.L.; Kuchroo, J.R.; Sage, P.T.; Liang, D.; Francisco, L.M.; Buck, J.; Thaker, Y.R.; Zhang, Q.; McArdel, S.L.; Juneja, V.R.; et al. PD-1 restraint of regulatory T cell suppressive activity is critical for immune tolerance. J. Exp. Med. 2021, 218, 218.
- Arce Vargas, F.; Furness, A.J.S.; Solomon, I.; Joshi, K.; Mekkaoui, L.; Lesko, M.H.; Miranda Rota, E.; Dahan, R.; Georgiou, A.; Sledzinska, A.; et al. Fc-Optimized Anti-CD25 depletes tumor-infiltrating regulatory T cells and synergizes with PD-1 blockade to eradicate established tumors. Immunity 2017, 46, 577–586.
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167.
- Asano, T.; Meguri, Y.; Yoshioka, T.; Kishi, Y.; Iwamoto, M.; Nakamura, M.; Sando, Y.; Yagita, H.; Koreth, J.; Kim, H.T.; et al. PD-1 modulates regulatory T-cell homeostasis during low-dose interleukin-2 therapy. Blood 2017, 129, 2186–2197.
- Huang, R.Y.; Francois, A.; McGray, A.R.; Miliotto, A.; Odunsi, K. Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. Oncoimmunology 2017, 6, e1249561.
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Aref, A.R.; Skoulidis, F.; Herter-Sprie, G.S.; Buczkowski, K.A.; Liu, Y.; Awad, M.M.; Denning, W.L.; et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. 2016, 76, 999–1008.
- Maj, T.; Wang, W.; Crespo, J.; Zhang, H.; Wei, S.; Zhao, L.; Vatan, L.; Shao, I.; Szeliga, W.; Lyssiotis, C.; et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 2017, 18, 1332–1341.
- Vigano, S.; Alatzoglou, D.; Irving, M.; Ménétrier-Caux, C.; Caux, C.; Romero, P.; Coukos, G. Targeting adenosine in cancer immunotherapy to enhance T-cell function. Front. Immunol. 2019, 10, 925.
- Koszałka, P.; Gołuńska, M.; Urban, A.; Stasiłojć, G.; Stanisławowski, M.; Majewski, M.; Składanowski, A.C.; Bigda, J. Specific activation of A3, A2A and A1 adenosine receptors in CD73-Knockout mice affects B16F10 melanoma growth, neovascularization, angiogenesis and macrophage infiltration. PLoS ONE 2016, 11, e0151420.
- Sorrentino, C.; Morello, S. Role of adenosine in tumor progression: Focus on A2b receptor as potential therapeutic target. J. Cancer Metastasis Treat. 2017, 3, 127–138.
- Frelaut, M.; Le Tourneau, C.; Borcoman, E. Hyperprogression under immunotherapy. Int. J. Mol. Sci. 2019, 20, 2674.
- Kim, C.G.; Kim, K.H.; Pyo, K.H.; Xin, C.F.; Hong, M.H.; Ahn, B.C.; Kim, Y.; Choi, S.J.; Yoon, H.I.; Lee, J.G.; et al. Hyperprogressive disease during PD-1/PD-L1 blockade in patients with non-small-cell lung cancer. Ann. Oncol. 2019, 30, 1104–1113.
- Saâda-Bouzid, E.; Defaucheux, C.; Karabajakian, A.; Coloma, V.P.; Servois, V.; Paoletti, X.; Even, C.; Fayette, J.; Guigay, J.; Loirat, D.; et al. Hyperprogression during anti-PD-1/PD-L1 therapy in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann. Oncol. 2017, 28, 1605–1611.
- Wang, X.; Wang, F.; Zhong, M.; Yarden, Y.; Fu, L. The biomarkers of hyperprogressive disease in PD-1/PD-L1 blockage therapy. Mol. Cancer 2020, 19, 81.