The tricarboxylic acid cycle (TCA) is a series of chemical reactions used in aerobic organisms to generate energy via the oxidation of acetylcoenzyme A (CoA) derived from carbohydrates, fatty acids and proteins.
- Krebs cycle
- tricarboxylic acid cycle
- cellular immunity
1. Introduction
The tricarboxylic acid cycle (TCA, also known as the Krebs cycle or the citric acid cycle) is a series of chemical reactions used in aerobic organisms (pro- and eukaryotes) to generate energy via the oxidation of acetyl-coenzyme A (CoA) derived from carbohydrates, fatty acids and proteins. In the eukaryotic system, the TCA cycle occurs exclusively in the matrix of the mitochondria, which are surrounded by two lipid bilayers and impermeable to polar solutes. The mitochondrial membrane separates energy-producing reactions from energy-consuming ones and retains the TCA cycle intermediates, which are small, highly polar, and electrophilic organic carboxylic acids, inside mitochondria. The intermediates of the TCA cycle take several roles as metabolites in metabolisms, such as insulin secretion and fatty acid synthesis (FAS) within mitochondria. In addition, they can be transported out of mitochondria through various shuttle mechanisms to participate in various biological processes. Of note, increasing evidence suggests that the TCA cycle intermediates play crucial roles in regulating cellular immunity. In cell stress conditions, the mitochondrial membrane may be disrupted and release the TCA cycle intermediates into the cytosol, thereby impacting the cellular immunity (Figure 1).
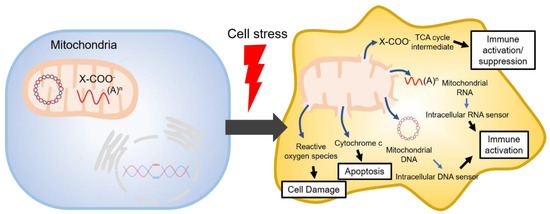
Figure 1. Mitochondrial danger signals in cell stress.
2. TCA Cycle Intermediates
2.1. Citrate
Citrate is formed in the TCA cycle by the condensation of oxaloacetate and acetyl-CoA, which is catalyzed by citrate synthase. Of note, the cellular levels of citrate are increased in pro-inflammatory macrophages [1][2][3]. Mitochondrial citrate can inhibit pyruvate dehydrogenase (PDH) and succinate dehydrogenase (SDH) at high concentrations [3]. PDH converts pyruvate to acetyl-CoA and CO2, whereas SDH oxidizes succinate to fumarate in the TCA cycle. Thereby, PDH and SDH catalyze two crucial steps of the TCA cycle, of which the first is the initiation of the TCA cycle and the second the generation of FADH2. Overall, citrate was found to exert inhibitory effects on the TCA cycle promoting ATP-consuming pathways [3][4][5].
In pro-inflammatory macrophages, mitochondrial citrate may be transported to the cytosol by SLC25A1 and converted to acetyl-CoA by ACLY [6][7]. SLC25A1 is upregulated in pro-inflammatory macrophages in a nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) or signal transducer and transcription (STAT)-dependent manner [2][3][6][7][8]. Conversely, the inhibition of SLC25A1 leads to the reduction of inflammatory mediators [7]. In line with this, the export and breakdown of mitochondrial citrate have been linked to the production of several important pro-inflammatory mediators in macrophages, such as nitric oxide (NO), ROS, and prostaglandin E2 (PGE2) [2][3][6][8][9]. The decrease in PGE2 production may be due to a decreased availability of citrate as adding exogenous acetate rescues the effect of SLC25A1 inhibition on PGE2 production, suggesting that the cytosolic citrate is an important signal molecule in inflammation [3]. Studies have demonstrated that the upregulation of the ACLY gene precedes SLC25A1 activation in both resting and pro-inflammatory macrophages, although SLC25A1 might be essential for providing the substrate for ACLY activity [6][8]. It is also interesting to note that SLC25A1 mutations that inactivate the citrate export are associated with severe mitochondrial dysfunction [2][10].
2.2. cis-Aconitate
cis-Aconitate is a TCA cycle intermediate, which is formed during the reversible transformation of citrate to isocitrate by the action of the enzyme aconitase (also known as aconitate hydratase). In this process, cis-aconitate normally does not dissociate from the active site of aconitase. Thus, not much is known about the cellular functions of cis-aconitate. Interestingly, it was recently reported that mitochondrial aconitase is associated with neurodegenerative disorders [11]. Congruently, an earlier study showed that lymphocytic mitochondrial aconitase activity is reduced in Alzheimer’s disease and mild cognitive impairment, implicating its significance in cellular homeostasis and immune cell functions [12]. In recent years, cis-aconitate has attracted a lot of attention because it was identified as the precursor of itaconate, which is formed from cis-aconitate through the decarboxylation catalyzed by the immune-responsive gene 1 (IRG1). Itaconate is upregulated upon immune activation, e.g., in response to LPS activation in macrophages, and has strong immunosuppressive properties. As the native itaconate is cell-impermeable, researchers often used structurally modified itaconate derivates with increased intracellular penetrance (e.g., dimethyl itaconate (DMI) and 4-octylitaconate) to study the intracellular functions of itaconate. So far, studies have shown that: (1) native and derivatized itaconate inhibits SDH, a component of both the TCA cycle and the Complex II of the electron transport chain (ETC) [13][14][15] consequently impairing immune activation (e.g., the hypoxia-induced factor (HIF)-1α-IL-1β axis) [14] and viral replication [15][16]; (2) activates the anti-inflammatory nuclear factor erythroid 2-related factor 2 (NRF2; also known as NFE2L2) pathway via alkylation of the Kelch-like ECH-associated protein 1 (KEAP1) protein [17][18]; and (3) regulates the translation of IκBζ and the subsequent activation of the cyclic AMP-dependent transcription factor (ATF3) albeit without affecting the primary transcriptional responses [19]. Collectively, previous studies have highlighted the immunosuppressive properties of itaconate and suggested that itaconate is an integral part of a negative-feedback loop in toll-like receptor (TLR)-mediated immune cell activation [17]. Of note, the cellular response to itaconate may differ from cell type to cell type and may also depend on the type of stimulation, as studies showed that endogenous itaconate does not induce NRF2 activation in neurons upon viral infection [15]. So far, most of the analysis on the immunosuppressive effect of itaconate has been performed in LPS-activated macrophages, and we do not know whether itaconate has the same impact on immune signaling pathways in other cell types and/or with different stimuli. Furthermore, it is to note that the most studied itaconate derivative is DMI, which is more electrophilic than the native form of itaconate. It is still unclear whether the native itaconate acts in the same way as its more electrophilic derivatives and deserves further investigations in the future.
2.3. Isocitrate
As mentioned above, isocitrate is formed in the TCA cycle by the isomerization of citrate catalyzed by aconitase. Studies have demonstrated an increase in citrate and a decrease in isocitrate in LPS-activated pro-inflammatory macrophages [1][6]. Furthermore, pro-inflammatory macrophages downregulate isocitrate dehydrogenase (IDH), resulting in an increased isocitrate:α-ketoglutarate ratio. Pro-inflammatory dendritic cells (DCs) and macrophages show increased glycolytic flux and a break in the TCA cycle, where pyruvate derived from glucose feeds into the TCA cycle but cannot continue beyond citrate/isocitrate. [3][20][21]. Generally, the cytosolic isoenzyme IDH1 is suppressed in cells activated by pro-inflammatory stimuli [3][22]. In glucose-deprived conditions, IDH1 mRNA expression is increased in pro-inflammatory macrophages [3][23]. This may explain why the production of NO and ROS is decreased in glucose-deprived cells. So far, the immunological roles of cytosolic and mitochondrial iso-citrate remain elusive and deserve future investigations.
2.4. α-Ketoglutarate
α-Ketoglutarate is formed in the TCA cycle by oxidative decarboxylation of iso-citrate catalyzed by IDH. α-Ketoglutarate is involved in various metabolic pathways [24][25][26][27][28]. In humans, IDH exists in three isoforms: IDH1, -2, and -3. IDH1 acts in the cytoplasm and, thus, outside the context of TCA, while IDH2 and -3, as part of the TCA cycle, are found within the mitochondrial matrix [29]. α-Ketoglutarate can also be generated from glutamine and glutamate by deamination to feed into the TCA cycle [30][31], whereas α-ketoglutarate can conversely serve as a precursor of glutamine, which is a nutritionally semi-essential amino acid [32]. Previous studies have identified immunosuppressive roles for α-ketoglutarate. Similarly, L-2-hydroxyglutarate, a derivative of α-ketoglutarate, was also shown to be immunosuppressive [33][34]. In this context, it is worth noting that mutations in IDH1 are associated with chronic inflammation [35][36]. IL-4-induced anti-inflammatory macrophages have been shown to accumulate the metabolite α-ketoglutarate [37]. It was also found that α-ketoglutarate suppresses the activation of pro-inflammatory macrophages (e.g., IL-1β expression) and supports endotoxin tolerance after activation [38][39]. α-Ketoglutarate regulates the pro-inflammatory NF-κB signaling by stimulating prolyl hydroxylase (PHD; a family of α-ketoglutarate-dependent dioxygenases (α-KGDD)). PHD1 inactivates IKK-β kinase via hydroxylation of Pro191, which, in turn, phosphorylates IκB-α, activating NF-κB [32][40]. α-Ketoglutarate inhibits stabilization of HIF-1α, a transcription factor inducing expression of immune-related genes (e.g., pro-inflammatory cytokines [41], glycolytic enzymes, and glucose transporters [42][43][44][45]) by donating an electron to HIF-specific PHD for prolyl hydroxylation leading to the degradation of HIF-1α [1][28][37][42][46][47][48][49][50]. In line with this, reduced levels of α-ketoglutarate were found to abolish PHD activity [51][28][49][52][53]. Besides, α-ketoglutarate is an important co-factor for the α-KGDD family of Jumonji-C-domain-containing histone demethylases (JMJDs) and the ten-eleven translocation (TET) proteins, which are respectively involved in histone and DNA demethylation [32][38][54][55][56][57][58][59][60]. Of note, histone demethylation is also associated with HIF-1α degradation [59][61]. α-Ketoglutarate facilitates macrophage polarization also via induction of fatty acid oxidation [40][48]. In addition, α-ketoglutarate produced by glutaminolysis regulates polarization of macrophages toward anti-inflammatory phenotypes, e. g., via JMJD3-mediated epigenetic changes [32][38][39][59]. Furthermore, glutamine anaplerosis (replenishing of TCA cycle intermediates from glutamate) represents an important metabolic module governing the polarization of macrophages towards an anti-inflammatory phenotype, e.g., in response to IL-4 [38][60][62]. Like most of the TCA cycle intermediates, α-ketoglutarate is transported from the mitochondrial matrix to the cytoplasm and vice versa. It crosses the outer mitochondrial membrane through the voltage-dependent anion channel (VDAC), and the inner mitochondrial membrane through the α-ketoglutarate/malate antiporter [32][63]. Obviously, α-ketoglutarate leaking from dysfunctioning mitochondria may exert similar anti-inflammatory functions as mitochondrial α-ketoglutarate transported into the cytosol.
2.5. Succinyl-CoA
Succinyl-CoA is formed by the decarboxylation of α-ketoglutarate catalyzed by α-ketoglutarate dehydrogenase (α-KGDH), a rate-limiting metabolic enzyme in the flux of the TCA cycle [54][64]. α-KGDH is responsive and sensitive to the levels of ROS impairing its function [54][65]. Of note, the increased α-KGDH activity in LPS-activated macrophages has been shown to limit the production of anti-inflammatory cytokine IL-10 [39][54]. An increase of cytosolic calcium leading to rapid mitochondrial acidification promotes α-KGDH activity, boosting NADH production and oxidative metabolism [54][66][67]. Succinyl-CoA accumulation can result in lysine succinylation of cellular proteins [1][68][69]. This post-translational modification (PTM) induces a 100-Da change in mass and masks the positive charge of the lysine side chain, likely resulting in a significant conformational change in the target protein [70]. While succinylation is largely considered to occur spontaneously, desuccinylation is at least partly enzymatic, e.g., via mitochondrial sirtuin 5 (SIRT5) [71]. LPS induces succinylation, which impacts macrophage function. LPS-induced hyper-succinylation of lysine 311 on pyruvate kinase M2 (PKM2), a key glycolytic enzyme interacting with HIF-1α and regulating aerobic glycolysis in macrophages, inhibits its pyruvate kinase activity by promoting its tetramer-to-dimer transition and induces IL-1β secretion [37][70][72][73]. LPS decreases the expression of the desuccinylase SIRT5 [1][60]. SIRT5 desucinylates various metabolic enzymes, including carbamoyl phosphate synthase 1 [60][74], SDH, PDH [60][70], acyl-CoA oxidase 1 [60][75], 3-hydroxy-3-methylglutaryl-CoA synthase 2 [60][76]. SIRT5 also desuccinylates PKM2 to prevent its entry into the nucleus and the formation of a HIF-1α/PKM2 complex [72]. Of note, the importance of this mechanism was highlighted by elevated levels of IL-1β in SIRT5-deficient mice examined in a colitis model [60][72]. Dysregulated succinate metabolism can also result in the accumulation of succinyl-CoA and lysine succinylation, a recently identified PTM [1][38][70]. Succinylation of certain proteins is found to drive immunosuppression in pro-inflammatory macrophages. Thus, the inflammatory effects of succinylation may be context-dependent and inducing various immunologic responses [38][37][77].
2.6. Succinate
Succinate is formed in the TCA cycle via the hydrolytic release of CoA catalyzed by succinyl-CoA synthetase. Alternatively, succinate is generated through the gamma-aminobutyric acid (GABA) shunt (a bypass of the TCA cycle, in which glutamine is used for the synthesis of glutamate, GABA, succinic semialdehyde, and eventually succinate) [1][43]. Succinate can be transported from the mitochondria to the cytosol by the dicarboxylic acid transporter. Succinate has been identified as an important metabolite in both host and microbial processes. Succinate accumulates in certain pathophysiological situations, particularly in areas of inflammation and metabolic stress [78][79]. Several studies have indicated its role of succinate in various cellular processes, such as epigenetic regulation, tumorigenesis, signal transduction, inflammation, and paracrine modulation [7][68][80][81] and have identified succinate as a metabolic signal governing local stress, tissue damage, and immunologic danger [1][82][83][84][85].
2.7. Fumarate
Fumarate is formed in the TCA cycle by the SDH-catalyzed dehydrogenation of succinate [78][86][87][88]. Activation of innate immune cells, such as macrophages and monocytes, with pro-inflammatory stimuli (e.g., LPS, IFN-γ) leads to remodeling of the TCA cycle and intracellular accumulation of fumarate [1][14][89][90][91][92]. LPS-activated macrophages show a substantial increase in flux through the induction of aspartate-arginosuccinate shunt, which fuels and maintains the TCA cycle flux [20][91]. Furthermore, being connected to the TCA cycle at fumarate, the induced aspartate-arginosuccinate shunt causes the accumulation of fumarate in activated macrophages [91]. Interestingly, the inhibition of the aspartate-arginosuccinate shunt leads to the downregulation of the pro-inflammatory mediators, including NO and IL-6, along with suppression of the ‘immunologic Warburg effect’ in activated macrophages [91]. Therefore, replenishing fumarate seems to be important for metabolic reprogramming of macrophages during inflammation [91]. The elevated cytokine response in these cells is linked to an increase in the amount of fumarate sufficient to drive the response by activating HIF-1α [93][94]. The accumulation of fumarate by inhibiting fumarate hydratase (FH; also known as fumarase) with FH inhibitor 1 (FHIN1) impairs cell death and reduces the formation of gasdermin D, the executioner of pyroptosis [90]. The in vivo-compatible FHIN2 elevates FH levels in vivo, while reducing IL-1β, indicating that fumarate inhibits pyroptosis in vivo [90]. Loss of function of FH in cancers increases fumarate levels leading to elevated accumulation of NRF2 [95][96]. Fumarate can attenuate autoimmune diseases, in part, through modulation of DC maturation and antigen-presenting capacity [97][98][99][100]. In general, fumarate is considered anti-inflammatory. Like other TCA cycle metabolites, fumarate is a cell-impermeable polar molecule. Therefore, researchers developed fumarate derivatives with increased intracellular penetrance to study immunological features of fumarate. Of note, the stable derivative of fumarate (DMF, trade name: Tecfidera®), is known to be a strong immunosuppressive. DMF is a leading oral therapy for multiple sclerosis. DMF is more electrophilic than fumarate and other structurally similar metabolites, such as DMI, and itaconate [19]. Therefore, whether the native fumarate has a similar effect in vivo and in vitro has yet to be determined [87][101]. Whereas the precise mechanism of immunosuppression has remained elusive, increasing evidence indicates that the critical mechanism of DMF is related to its high electrophilicity, which allows DMF to modify proteins by reacting with cysteines via an electrophilic (Michael-type) addition [102][103], impacting the function of proteins. Several groups have proposed that DMF stimulates an antioxidant response by modifying cysteine residues in the NRF2-KEAP1 complex, which is a major electrophile response pathway in mammals [87][95][104][105][106][107][108]. The transcription factor NRF2 has been shown to alleviate inflammation by inducing heme oxygenase-1 (HO-1), which potentially increases IRG1 and downregulates TNF-α expression [108][109][110][111][112]. An NRF2 deficiency causes an exacerbation of inflammation in a variety of murine models, such as sepsis, pleurisy, and emphysema [113][114][115][116], as well as autoimmune phenotypes in some murine strains [117][118]. GAPDH has been hypothesized to play a key role in the mechanism of action of DMF [119]. DMF has been suggested to modify the active site cysteine (Cys152 in humans) and to inactivate GAPDH. Thereby, DMF may downregulate glycolysis, ultimately causing immunosuppression [119]. However, a recent study suggested that this mechanism does not have the primary role in DMF-mediated immunosuppression [90].
2.8. Malate and Oxaloacetate
L-Malate is formed by the hydration of fumarate catalyzed by FH. In the last reaction of the TCA cycle, L-malate dehydrogenase oxidizes L-malate to oxaloacetate. Not much is known about the immunological role of intracellular L-malate and oxaloacetate, although the activation of macrophages with LPS results in increased levels of L-malate [1][42]. Oxaloacetate serves as a strong inhibitor of SDH, thereby limiting post-ischemic oxidation of succinate. It would be worth examining whether the endogenously formed oxaloacetate conveys protection through reduced SDH activity during reperfusion [120]. Future studies are needed to determine the immunological roles of L-malate and oxaloacetate during a mitochondrial disruption.
This entry is adapted from the peer-reviewed paper 10.3390/life11010069
References
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242.
- Infantino, V.; Iacobazzi, V.; Menga, A.; Avantaggiati, M.L.; Palmieri, F. A key role of the mitochondrial citrate carrier (SLC25A1) in TNFα- and IFNγ-triggered inflammation. Biochim. Biophys. Acta 2014, 1839, 1217–1225.
- Williams, N.C.; O’Neill, L.A.J. A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front. Immunol. 2018, 9, 141.
- Hillar, M.; Lott, V.; Lennox, B. Correlation of the effects of citric acid cycle metabolites on succinate oxidation by rat liver mitochondria and submitochondrial particles. J. Bioenergy 1975, 7, 1–16.
- Taylor, W.M.; Halperin, M.L. Regulation of pyruvate dehydrogenase in muscle. Inhibition by citrate. J. Biol. Chem. 1973, 248, 6080–6083.
- Infantino, V.; Convertini, P.; Cucci, L.; Panaro, M.A.; Di Noia, M.A.; Calvello, R.; Palmieri, F.; Iacobazzi, V. The mitochondrial citrate carrier: A new player in inflammation. Biochem. J. 2011, 438, 433–436.
- Mills, E.; O’Neill, L.A. Succinate: A metabolic signal in inflammation. Trends Cell Biol. 2014, 24, 313–320.
- Infantino, V.; Iacobazzi, V.; Palmieri, F.; Menga, A. ATP-citrate lyase is essential for macrophage inflammatory response. Biochem. Biophys. Res. Commun. 2013, 440, 105–111.
- Conti, M.G.; Angelidou, A.; Diray-Arce, J.; Smolen, K.K.; Lasky-Su, J.; De Curtis, M.; Levy, O. Immunometabolic approaches to prevent, detect, and treat neonatal sepsis. Pediatric Res. 2020, 87, 399–405.
- Iacobazzi, V.; Infantino, V. Citrate—New functions for an old metabolite. Biol. Chem. 2014, 395, 387–399.
- Khodagholi, F.; Shaerzadeh, F.; Montazeri, F. Mitochondrial Aconitase in Neurodegenerative Disorders: Role of a Metabolism- related Molecule in Neurodegeneration. Curr. Drug Targets 2018, 19, 973–985.
- Mangialasche, F.; Baglioni, M.; Cecchetti, R.; Kivipelto, M.; Ruggiero, C.; Piobbico, D.; Kussmaul, L.; Monastero, R.; Brancorsini, S.; Mecocci, P. Lymphocytic mitochondrial aconitase activity is reduced in Alzheimer’s disease and mild cognitive impairment. J. Alzheimer’s Dis. 2015, 44, 649–660.
- Cordes, T.; Wallace, M.; Michelucci, A.; Divakaruni, A.S.; Sapcariu, S.C.; Sousa, C.; Koseki, H.; Cabrales, P.; Murphy, A.N.; Hiller, K.; et al. Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. J. Biol. Chem. 2016, 291, 14274–14284.
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166.
- Daniels, B.P.; Kofman, S.B.; Smith, J.R.; Norris, G.T.; Snyder, A.G.; Kolb, J.P.; Gao, X.; Locasale, J.W.; Martinez, J.; Gale, M., Jr.; et al. The Nucleotide Sensor ZBP1 and Kinase RIPK3 Induce the Enzyme IRG1 to Promote an Antiviral Metabolic State in Neurons. Immunity 2019, 50, 64–76.e4.
- Minton, K. Neuronal itaconate restricts viral infection. Nat. Rev. Immunol. 2019, 19, 67.
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117.
- Tang, C.; Wang, X.; Xie, Y.; Cai, X.; Yu, N.; Hu, Y.; Zheng, Z. 4-Octyl Itaconate Activates Nrf2 Signaling to Inhibit Pro-Inflammatory Cytokine Production in Peripheral Blood Mononuclear Cells of Systemic Lupus Erythematosus Patients. Cell Physiol. Biochem. 2018, 51, 979–990.
- Bambouskova, M.; Gorvel, L.; Lampropoulou, V.; Sergushichev, A.; Loginicheva, E.; Johnson, K.; Korenfeld, D.; Mathyer, M.E.; Kim, H.; Huang, L.H.; et al. Electrophilic properties of itaconate and derivatives regulate the IkappaBzeta-ATF3 inflammatory axis. Nature 2018, 556, 501–504.
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430.
- Everts, B.; Amiel, E.; Huang, S.C.; Smith, A.M.; Chang, C.H.; Lam, W.Y.; Redmann, V.; Freitas, T.C.; Blagih, J.; van der Windt, G.J.; et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKɛ supports the anabolic demands of dendritic cell activation. Nat. Immunol. 2014, 15, 323–332.
- Haseeb, A.; Makki, M.S.; Haqqi, T.M. Modulation of ten-eleven translocation 1 (TET1), Isocitrate Dehydrogenase (IDH) expression, α-Ketoglutarate (α-KG), and DNA hydroxymethylation levels by interleukin-1β in primary human chondrocytes. J. Biol. Chem. 2014, 289, 6877–6885.
- Palmieri, E.M.; Spera, I.; Menga, A.; Infantino, V.; Porcelli, V.; Iacobazzi, V.; Pierri, C.L.; Hooper, D.C.; Palmieri, F.; Castegna, A. Acetylation of human mitochondrial citrate carrier modulates mitochondrial citrate/malate exchange activity to sustain NADPH production during macrophage activation. Biochim. Biophys. Acta 2015, 1847, 729–738.
- Bruns, G.A.; Eisenman, R.E.; Gerald, P.S. Human mitochondrial NADP-dependent isocitrate dehydrogenase in man-mouse somatic cell hybrids. Cytogenet. Cell Genet. 1976, 17, 200–211.
- Shimizu, N.; Giles, R.E.; Kucherlapati, R.S.; Shimizu, Y.; Ruddle, F.H. Somatic cell genetic assignment of the human gene for mitochondrial NADP-linked isocitrate dehydrogenase to the long arm of chromosome 15. Somat. Cell Genet. 1977, 3, 47–60.
- Geisbrecht, B.V.; Gould, S.J. The human PICD gene encodes a cytoplasmic and peroxisomal NADP(+)-dependent isocitrate dehydrogenase. J. Biol. Chem. 1999, 274, 30527–30533.
- Corpas, F.J.; Barroso, J.B.; Sandalio, L.M.; Palma, J.M.; Lupiáñez, J.A.; del Río, L.A. Peroxisomal NADP-Dependent Isocitrate Dehydrogenase. Characterization and Activity Regulation during Natural Senescence. Plant Physiol. 1999, 121, 921–928.
- Xiang, S.; Gu, H.; Jin, L.; Thorne, R.F.; Zhang, X.D.; Wu, M. LncRNA IDH1-AS1 links the functions of c-Myc and HIF1α via IDH1 to regulate the Warburg effect. Proc. Natl. Acad. Sci. USA 2018, 115, E1465–E1474.
- Tommasini-Ghelfi, S.; Murnan, K.; Kouri, F.M.; Mahajan, A.S.; May, J.L.; Stegh, A.H. Cancer-associated mutation and beyond: The emerging biology of isocitrate dehydrogenases in human disease. Sci. Adv. 2019, 5, eaaw4543.
- Chen, L.; Chen, X.W.; Huang, X.; Song, B.L.; Wang, Y. Regulation of glucose and lipid metabolism in health and disease. Sci. China Life Sci. 2019, 62, 1420–1458.
- Song, W.; Li, D.; Tao, L.; Luo, Q.; Chen, L. Solute carrier transporters: The metabolic gatekeepers of immune cells. Acta Pharm Sin. B 2020, 10, 61–78.
- Abla, H.; Sollazzo, M.; Gasparre, G.; Iommarini, L.; Porcelli, A.M. The multifaceted contribution of α-ketoglutarate to tumor progression: An opportunity to exploit? Semin. Cell Dev. Biol. 2020, 98, 26–33.
- Weinberg, S.E.; Singer, B.D.; Steinert, E.M.; Martinez, C.A.; Mehta, M.M.; Martínez-Reyes, I.; Gao, P.; Helmin, K.A.; Abdala-Valencia, H.; Sena, L.A.; et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 2019, 565, 495–499.
- Xu, T.; Stewart, K.M.; Wang, X.; Liu, K.; Xie, M.; Ryu, J.K.; Li, K.; Ma, T.; Wang, H.; Ni, L.; et al. Metabolic control of TH17 and induced Treg cell balance by an epigenetic mechanism. Nat. Cell Biol. 2017, 548, 228–233.
- Hartman, D.J.; Binion, D.; Regueiro, M.; Schraut, W.; Bahary, N.; Sun, W.; Nikiforova, M.; Pai, R.K. Isocitrate dehydrogenase-1 is mutated in inflammatory bowel disease-associated intestinal adenocarcinoma with low-grade tubuloglandular histology but not in sporadic intestinal adenocarcinoma. Am. J. Surg. Pathol. 2014, 38, 1147–1156.
- Auezova, R.; Ivanova, N.; Akshulakov, S.; Zhetpisbaev, B.; Kozhakhmetova, A.; Ryskeldiyev, N.; Mustafin, K.; Teltayev, D.; Auezova, L. Isocitrate dehydrogenase 1 mutation is associated with reduced levels of inflammation in glioma patients. Cancer Manag. Res. 2019, 11, 3227–3236.
- Harber, K.J.; de Goede, K.E.; Verberk, S.G.S.; Meinster, E.; de Vries, H.E.; van Weeghel, M.; de Winther, M.P.J.; Van den Bossche, J. Succinate Is an Inflammation-Induced Immunoregulatory Metabolite in Macrophages. Metabolites 2020, 10, 372.
- Ryan, D.G.; O’Neill, L.A.J. Krebs Cycle Reborn in Macrophage Immunometabolism. Annu. Rev. Immunol. 2020, 38, 289–313.
- Liu, P.S.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; Di Conza, G.; Cheng, W.C.; Chou, C.H.; Vavakova, M.; et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017, 18, 985–994.
- Littlewood-Evans, A.; Sarret, S.; Apfel, V.; Loesle, P.; Dawson, J.; Zhang, J.; Muller, A.; Tigani, B.; Kneuer, R.; Patel, S.; et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J. Exp. Med. 2016, 213, 1655–1662.
- Cummins, E.P.; Berra, E.; Comerford, K.M.; Ginouves, A.; Fitzgerald, K.T.; Seeballuck, F.; Godson, C.; Nielsen, J.E.; Moynagh, P.; Pouyssegur, J.; et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc. Natl. Acad. Sci. USA 2006, 103, 18154–18159.
- Galván-Peña, S.; O’Neill, L.A. Metabolic reprograming in macrophage polarization. Front. Immunol. 2014, 5, 420.
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462.
- Wang, T.; Liu, H.; Lian, G.; Zhang, S.Y.; Wang, X.; Jiang, C. HIF1 α-Induced Glycolysis Metabolism Is Essential to the Activation of Inflammatory Macrophages. Mediat. Inflamm. 2017, 2017, 9029327.
- Van Uden, P.; Kenneth, N.S.; Rocha, S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem. J. 2008, 412, 477–484.
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008, 453, 807–811.
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773.
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744.
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Habdank, M.; Krönke, J.; Bullinger, L.; Späth, D.; Kayser, S.; Zucknick, M.; Götze, K.; et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J. Clin. Oncol. 2010, 28, 3636–3643.
- MacKenzie, E.D.; Selak, M.A.; Tennant, D.A.; Payne, L.J.; Crosby, S.; Frederiksen, C.M.; Watson, D.G.; Gottlieb, E. Cell-permeating alpha-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol. Cell. Biol. 2007, 27, 3282–3289.
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell. 2010, 40, 294–309.
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102.
- Bargiela, D.; Burr, S.P.; Chinnery, P.F. Mitochondria and Hypoxia: Metabolic Crosstalk in Cell-Fate Decisions. Trends Endocrinol. Metab. 2018, 29, 249–259.
- Lio, C.J.; Huang, S.C. Circles of Life: Linking metabolic and epigenetic cycles to immunity. Immunology 2020, 161, 165–174.
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935.
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303.
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307.
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012, 26, 1326–1338.
- Tsukada, Y.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816.
- Zasłona, Z.; O’Neill, L.A.J. Cytokine-like Roles for Metabolites in Immunity. Mol. Cell 2020, 78, 814–823.
- Peruzzotti-Jametti, L.; Pluchino, S. Targeting Mitochondrial Metabolism in Neuroinflammation: Towards a Therapy for Progressive Multiple Sclerosis. Trends Mol. Med. 2018, 24, 838–855.
- Palsson-McDermott, E.M.; O’Neill, L.A. The Warburg effect then and now: From cancer to inflammatory diseases. Bioessays 2013, 35, 965–973.
- Monné, M.; Miniero, D.V.; Iacobazzi, V.; Bisaccia, F.; Fiermonte, G. The mitochondrial oxoglutarate carrier: From identification to mechanism. J. Bioenerg. Biomembr. 2013, 45, 1–13.
- Yokota, I.; Kuroda, Y. Alpha-ketoglutarate dehydrogenase complex. Nihon Rinsho. Jpn. J. Clin. Med. 2002, 60 (Suppl. S4), 130–132.
- McLain, A.L.; Szweda, P.A.; Szweda, L.I. α-Ketoglutarate dehydrogenase: A mitochondrial redox sensor. Free Radic. Res. 2011, 45, 29–36.
- Tretter, L.; Adam-Vizi, V. Alpha-ketoglutarate dehydrogenase: A target and generator of oxidative stress. Philos. Trans. R Soc. Lond. B Biol. Sci. 2005, 360, 2335–2345.
- Griffiths, E.J.; Rutter, G.A. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim. Biophys. Acta 2009, 1787, 1324–1333.
- Cho, E.H. Succinate as a Regulator of Hepatic Stellate Cells in Liver Fibrosis. Front. Endocrinol. 2018, 9, 455.
- Zhang, Y.; Bharathi, S.S.; Rardin, M.J.; Lu, J.; Maringer, K.V.; Sims-Lucas, S.; Prochownik, E.V.; Gibson, B.W.; Goetzman, E.S. Lysine desuccinylase SIRT5 binds to cardiolipin and regulates the electron transport chain. J. Biol. Chem. 2017, 292, 10239–10249.
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.; Skinner, M.E.; et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 2013, 50, 919–930.
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809.
- Wang, F.; Wang, K.; Xu, W.; Zhao, S.; Ye, D.; Wang, Y.; Xu, Y.; Zhou, L.; Chu, Y.; Zhang, C.; et al. SIRT5 Desuccinylates and Activates Pyruvate Kinase M2 to Block Macrophage IL-1β Production and to Prevent DSS-Induced Colitis in Mice. Cell Rep. 2017, 19, 2331–2344.
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.; et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015, 21, 65–80.
- Nakagawa, T.; Lomb, D.J.; Haigis, M.C.; Guarente, L. SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell 2009, 137, 560–570.
- Chen, X.F.; Tian, M.X.; Sun, R.Q.; Zhang, M.L.; Zhou, L.S.; Jin, L.; Chen, L.L.; Zhou, W.J.; Duan, K.L.; Chen, Y.J.; et al. SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. 2018, 19, e45124.
- Rardin, M.J.; He, W.; Nishida, Y.; Newman, J.C.; Carrico, C.; Danielson, S.R.; Guo, A.; Gut, P.; Sahu, A.K.; Li, B.; et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 2013, 18, 920–933.
- Wu, J.Y.; Huang, T.W.; Hsieh, Y.T.; Wang, Y.F.; Yen, C.C.; Lee, G.L.; Yeh, C.C.; Peng, Y.J.; Kuo, Y.Y.; Wen, H.T.; et al. Cancer-Derived Succinate Promotes Macrophage Polarization and Cancer Metastasis via Succinate Receptor. Mol. Cell 2020, 77, 213–227.e5.
- Akram, M. Citric acid cycle and role of its intermediates in metabolism. Cell Biochem. Biophys. 2014, 68, 475–478.
- Connors, J.; Dawe, N.; Van Limbergen, J. The Role of Succinate in the Regulation of Intestinal Inflammation. Nutrients 2018, 11, 25.
- Ariza, A.C.; Deen, P.M.; Robben, J.H. The succinate receptor as a novel therapeutic target for oxidative and metabolic stress-related conditions. Front. Endocrinol. 2012, 3, 22.
- De Castro Fonseca, M.; Aguiar, C.J.; da Rocha Franco, J.A.; Gingold, R.N.; Leite, M.F. GPR91: Expanding the frontiers of Krebs cycle intermediates. Cell Commun. Signal. 2016, 14, 3.
- Toma, I.; Kang, J.J.; Sipos, A.; Vargas, S.; Bansal, E.; Hanner, F.; Meer, E.; Peti-Peterdi, J. Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. J. Clin. Investig. 2008, 118, 2526–2534.
- Keiran, N.; Ceperuelo-Mallafré, V.; Calvo, E.; Hernández-Alvarez, M.I.; Ejarque, M.; Núñez-Roa, C.; Horrillo, D.; Maymó-Masip, E.; Rodríguez, M.M.; Fradera, R.; et al. SUCNR1 controls an anti-inflammatory program in macrophages to regulate the metabolic response to obesity. Nat. Immunol. 2019, 20, 581–592.
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435.
- Rubic, T.; Lametschwandtner, G.; Jost, S.; Hinteregger, S.; Kund, J.; Carballido-Perrig, N.; Schwärzler, C.; Junt, T.; Voshol, H.; Meingassner, J.G.; et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat. Immunol. 2008, 9, 1261–1269.
- Jiang, S.; Yan, W. Succinate in the cancer-immune cycle. Cancer Lett. 2017, 390, 45–47.
- Ryan, D.G.; O’Neill, L.A.J. Krebs cycle rewired for macrophage and dendritic cell effector functions. FEBS Lett. 2017, 591, 2992–3006.
- King, A.; Selak, M.A.; Gottlieb, E. Succinate dehydrogenase and fumarate hydratase: Linking mitochondrial dysfunction and cancer. Oncogene 2006, 25, 4675–4682.
- Murphy, M.P.; O’Neill, L.A.J. Krebs Cycle Reimagined: The Emerging Roles of Succinate and Itaconate as Signal Transducers. Cell 2018, 174, 780–784.
- Humphries, F.; Shmuel-Galia, L.; Ketelut-Carneiro, N.; Li, S.; Wang, B.; Nemmara, V.V.; Wilson, R.; Jiang, Z.; Khalighinejad, F.; Muneeruddin, K.; et al. Succination inactivates gasdermin D and blocks pyroptosis. Science 2020, 369, 1633–1637.
- Patil, N.K.; Bohannon, J.K.; Hernandez, A.; Patil, T.K.; Sherwood, E.R. Regulation of leukocyte function by citric acid cycle intermediates. J. Leukoc. Biol. 2019, 106, 105–117.
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784.
- Infantino, V.; Convertini, P.; Cucci, L.; Panaro, M.A.; Di Noia, M.A.; Calvello, R.; Palmieri, F.; Iacobazzi, V. The mitochondrial citrate carrier: A new player in inflammation. Biochem. J. 2011, 438, 433–436.
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498.
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203.
- Tomlinson, I.P.; Alam, N.A.; Rowan, A.J.; Barclay, E.; Jaeger, E.E.; Kelsell, D.; Leigh, I.; Gorman, P.; Lamlum, H.; Rahman, S.; et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 2002, 30, 406–410.
- Hosseini, A.; Masjedi, A.; Baradaran, B.; Hojjat-Farsangi, M.; Ghalamfarsa, G.; Anvari, E.; Jadidi-Niaragh, F. Dimethyl fumarate: Regulatory effects on the immune system in the treatment of multiple sclerosis. J. Cell Physiol. 2019, 234, 9943–9955.
- Diebold, M.; Sievers, C.; Bantug, G.; Sanderson, N.; Kappos, L.; Kuhle, J.; Lindberg, R.L.P.; Derfuss, T. Dimethyl fumarate influences innate and adaptive immunity in multiple sclerosis. J. Autoimmun. 2018, 86, 39–50.
- Ghoreschi, K.; Brück, J.; Kellerer, C.; Deng, C.; Peng, H.; Rothfuss, O.; Hussain, R.Z.; Gocke, A.R.; Respa, A.; Glocova, I.; et al. Fumarates improve psoriasis and multiple sclerosis by inducing type II dendritic cells. J. Exp. Med. 2011, 208, 2291–2303.
- Peng, H.; Guerau-de-Arellano, M.; Mehta, V.B.; Yang, Y.; Huss, D.J.; Papenfuss, T.L.; Lovett-Racke, A.E.; Racke, M.K. Dimethyl fumarate inhibits dendritic cell maturation via nuclear factor κB (NF-κB) and extracellular signal-regulated kinase 1 and 2 (ERK1/2) and mitogen stress-activated kinase 1 (MSK1) signaling. J. Biol. Chem. 2012, 287, 28017–28026.
- Ashrafian, H.; Czibik, G.; Bellahcene, M.; Aksentijević, D.; Smith, A.C.; Mitchell, S.J.; Dodd, M.S.; Kirwan, J.; Byrne, J.J.; Ludwig, C.; et al. Fumarate is cardioprotective via activation of the Nrf2 antioxidant pathway. Cell Metab. 2012, 15, 361–371.
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692.
- Blewett, M.M.; Xie, J.; Zaro, B.W.; Backus, K.M.; Altman, A.; Teijaro, J.R.; Cravatt, B.F. Chemical proteomic map of dimethyl fumarate-sensitive cysteines in primary human T cells. Sci. Signal. 2016, 9, rs10.
- Brennan, M.S.; Matos, M.F.; Li, B.; Hronowski, X.; Gao, B.; Juhasz, P.; Rhodes, K.J.; Scannevin, R.H. Dimethyl fumarate and monoethyl fumarate exhibit differential effects on KEAP1, NRF2 activation, and glutathione depletion in vitro. PLoS ONE 2015, 10, e0120254.
- ElAzzouny, M.; Tom, C.T.; Evans, C.R.; Olson, L.L.; Tanga, M.J.; Gallagher, K.A.; Martin, B.R.; Burant, C.F. Dimethyl Itaconate Is Not Metabolized into Itaconate Intracellularly. J. Biol. Chem. 2017, 292, 4766–4769.
- Ooi, A.; Wong, J.C.; Petillo, D.; Roossien, D.; Perrier-Trudova, V.; Whitten, D.; Min, B.W.; Tan, M.H.; Zhang, Z.; Yang, X.J.; et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell 2011, 20, 511–523.
- Adam, J.; Hatipoglu, E.; O’Flaherty, L.; Ternette, N.; Sahgal, N.; Lockstone, H.; Baban, D.; Nye, E.; Stamp, G.W.; Wolhuter, K.; et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: Roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 2011, 20, 524–537.
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624.
- Konrad, F.M.; Knausberg, U.; Hone, R.; Ngamsri, K.C.; Reutershan, J. Tissue heme oxygenase-1 exerts anti-inflammatory effects on LPS-induced pulmonary inflammation. Mucosal Immunol. 2016, 9, 98–111.
- Zhao, C.; Jiang, P.; He, Z.; Yuan, X.; Guo, J.; Li, Y.; Hu, X.; Cao, Y.; Fu, Y.; Zhang, N. Dimethyl itaconate protects against lippolysacchride-induced mastitis in mice by activating MAPKs and Nrf2 and inhibiting NF-kappaB signaling pathways. Microb. Pathog. 2019, 133, 103541.
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322.
- Kobayashi, E.; Suzuki, T.; Yamamoto, M. Roles nrf2 plays in myeloid cells and related disorders. Oxid. Med. Cell. Longev. 2013, 2013, 529219.
- Itoh, K.; Mochizuki, M.; Ishii, Y.; Ishii, T.; Shibata, T.; Kawamoto, Y.; Kelly, V.; Sekizawa, K.; Uchida, K.; Yamamoto, M. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-Δ(12,14)-prostaglandin J(2). Mol. Cell Biol. 2004, 24, 36–45.
- Ishii, Y.; Itoh, K.; Morishima, Y.; Kimura, T.; Kiwamoto, T.; Iizuka, T.; Hegab, A.E.; Hosoya, T.; Nomura, A.; Sakamoto, T.; et al. Transcription factor Nrf2 plays a pivotal role in protection against elastase-induced pulmonary inflammation and emphysema. J. Immunol. 2005, 175, 6968–6975.
- Reisin, L.; Schneeweiss, A. Spontaneous disappearance of cough induced by angiotensin-converting enzyme inhibitors (captopril or enalapril). Am. J. Cardiol. 1992, 70, 398–399.
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006, 116, 984–995.
- Yoh, K.; Itoh, K.; Enomoto, A.; Hirayama, A.; Yamaguchi, N.; Kobayashi, M.; Morito, N.; Koyama, A.; Yamamoto, M.; Takahashi, S. Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney Int. 2001, 60, 1343–1353.
- Ma, Q.; Battelli, L.; Hubbs, A.F. Multiorgan autoimmune inflammation, enhanced lymphoproliferation, and impaired homeostasis of reactive oxygen species in mice lacking the antioxidant-activated transcription factor Nrf2. Am. J. Pathol. 2006, 168, 1960–1974.
- Kornberg, M.D.; Bhargava, P.; Kim, P.M.; Putluri, V.; Snowman, A.M.; Putluri, N.; Calabresi, P.A.; Snyder, S.H. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science 2018, 360, 449–453.
- Zuurbier, C.J.; Bertrand, L.; Beauloye, C.R.; Andreadou, I.; Ruiz-Meana, M.; Jespersen, N.R.; Kula-Alwar, D.; Prag, H.A.; Eric Botker, H.; Dambrova, M.; et al. Cardiac metabolism as a driver and therapeutic target of myocardial infarction. J. Cell. Mol. Med. 2020, 24, 5937–5954.